文章刊载于《临床实验室》杂志2023年6月刊“恶性肿瘤实验室检测”专题-「实验室诊断技术导航」版块 页码p96-p105
摘要:目的 本研究的目的是通过血清肠道菌群相关代谢物的检测用于诊断结直肠癌和腺瘤患者。方法 通过整合分析非靶向代谢组质谱检测获得的外周血代谢组数据和配对的粪便宏基因组测序数据获得在肠癌和腺瘤患者外周血中显著改变的肠道菌群相关代谢物,这些代谢物进一步通过靶向代谢组学验证用于肠癌诊断模型的建立。结果 共计885个血清代谢物在肠癌和腺瘤患者中其丰度显著改变,其中有8个肠道菌群相关代谢物可以被非靶向和靶向代谢物检测重复稳定的测定并用于肠癌患者的鉴别。基于这些肠道菌群相关代谢物建立的用于肠癌和腺瘤患者的诊断模型其曲线下面积为0.98 (95% CI 0.94 to 1.00),在验证队列其曲线下面积为0.92,敏感性和特异性分别为83.5%和84.9%,显著优于癌胚抗原用于肠癌诊断的曲线下面积(0.72)。而且,该模型用于早期肠癌和腺瘤的诊断的曲线下面积分别为0.93和0.84。结论 肠癌患者的肠道菌群重塑伴随着血清代谢组的改变,而且本研究发现肠道菌群相关代谢物可用于肠癌和腺瘤患者的检测。

结直肠癌(CRC)日益成为人们健康的重大挑战,肠癌的早期诊断是提高患者生存率的有效方式。目前几种用于肠癌检测的手段已经用于临床实践,例如非侵入性的粪便潜血试验和癌胚抗原检测,以及侵入性的肠镜检查。然而由于这些方法的低效率和侵入性限制了它们的广泛开展,因此发展无创且准确的检测方法尤为重要。
菌群对人类疾病特别是癌症的影响越来越受到关注,由于空间上的接近性,CRC的发生显著受到肠道菌群的影响。肠癌患者的菌群构成显著发生改变,例如Bacteroides, Parvimonas, Bilophila 和Fusobacteriu 丰度增高, Ruminococcus, Bifidobacterium 和Streptococcu 丰度降低。这些改变的菌群重塑肠道局部的免疫反应,产生基因毒素以及次级胆汁酸等菌群代谢物从而引发肠癌和促进肠癌发展。
癌症发生伴随着整体的代谢改变,影响癌组织及其微环境。这与基因组和蛋白组的改变相比,代谢组的改变更易被观察到,因此代谢物的改变成为较为理想的标志物。已有多项研究表明,肠道菌群产生的代谢物能够进入血液循环并且在远处器官呈现调控功能,它与肠癌相关的血清代谢物也已有较多研究,然而肠道菌群相关的菌群改变对外周血的影响,特别是其临床应用尚不明确。
本研究中,我们整合分析了血清代谢组和配对的粪便宏基因组数据,获得了一组可用于肠癌和腺瘤诊断的外周血肠道菌群相关代谢物,并基于这些代谢物建立了肠癌和腺瘤的诊断模型(图1. A)。
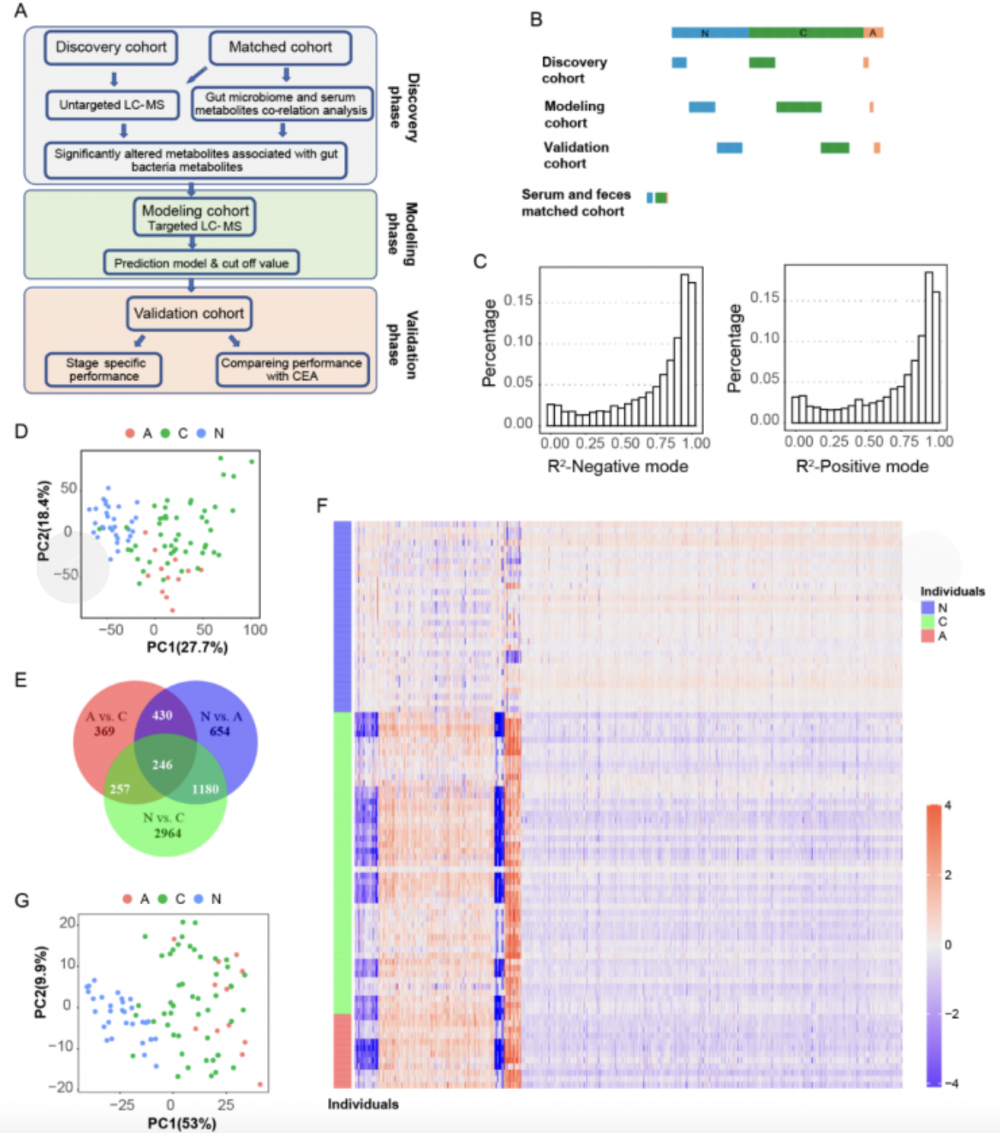
注:A:实验设计和分析流程图。发现阶段,血清和粪便样本配对队列进行非靶向代谢组和宏基因组测序,用于表明血清代谢物与肠道菌群相关而且肠癌和腺瘤患者外周血代谢物显著改变。发现队列进行靶向代谢组检测选择候选标志物,基于生物标志物组合建立诊断模型并进一步在验证队列进行验证。B:研究队列的构成。发现队列,建模队列以及验证队列分别采集患者血清。配对队列患者的粪便和血清同时采集用于关联研究。N:正常对照 (蓝色);A:腺瘤 (红色);C: 肠癌(绿色)。C:非靶向代谢检测的R2 值分布。D:主成分分析图表明血清代谢物在正常,腺瘤和肠癌患者间的差异。N:正常对照 (蓝色);A:腺瘤 (红色);C: 肠癌(绿色)。E:Venn 图表明各组之间代谢物分布情况。N:正常对照;A:腺瘤;C: 肠癌。F:热图表明代谢物在N:正常对照 (蓝色);A:腺瘤 (红色);C: 肠癌(绿色)各组之间的差异。G:基于各组之间明显差异的代谢物,主成分分析图表明血清代谢物在正常,腺瘤和肠癌患者间的差异。N:正常对照 (蓝色);A:腺瘤 (红色);C: 肠癌(绿色)。
图 1 肠癌和腺瘤患者血清代谢组的改变
研究对象与方法 1.研究对象:共计484例45至75岁的肠癌患者,腺瘤患者以及健康对照的血清样本用于本研究,其中44例肠癌患者的粪便样本也同时采集。分为四个研究队列;发现队列、建模队列、验证队列、血清和粪便样本匹配队列,每个队列中均有不同数量的正常人、腺瘤和肠癌 患者样本(图1.B)。肠癌及腺瘤患者的血清样本均为初次治疗前采集,用于整合分析的粪便样本也于初次治疗前采集。本研究经中国医学科学院肿瘤医院伦理委员会批准。 2.代谢物提取:靶向代谢物提取,240 μL 乙腈:异丙醇(3:1 体积比, Thermo Fisher),加入到60 μL 血清中。再加入甲酸铵 60μL (0.5 g/mL, Thermo Fisher)以及6 μL 内标液100 µg/mL 的L-Tyrosine-(phenyl-3,5-d2) (Sigma-Aldrich) 用于沉淀蛋白。10 µg/mL 的 13C-Cholic Acid (Cambridge Isotope Laboratories) 和 60 µg/mL 的 Doxercalciferol (MedChem Express)加入后涡旋 4 分钟,17949 g 离心5分钟之后, 200 μL 上清液移入另一个试管中并 - 60℃ 冻干。冻干物再以 75 μL 的 55% methanol (含有0.1% formic acid,Thermo Fisher)重悬。 3.非靶向代谢组检测:上述获得的代谢物采用Q Exactive mass spectrometer和 UltiMate3000 UPLC (Thermo Fisher) 进行检测,质谱检测数据采用Progenesis QI Software (Nonlinear Dynamics, Durham, NC, USA)进一步分析。 4.代谢物注释:MS-DIAL 4.24 程序用于代谢物注释。 5.粪便宏基因组测序:粪便样本DNA采用QIAamp DNA Stool Mini Kit (QIAGEN)提取,使用鸟枪法进行宏基因组测序,并进行肠道微生物组的分类和功能分析。 6.靶向代谢组检测:优化的Pseudotargeted 方法用于靶向代谢物的检测。 7.数据统计分析:数据的处理,统计分析,预测模型的建立采用 R programming (v3.6.1)。ANOVA with Tukey’s HSD test 用于数据统计分析,代谢物在各队列中的差异判定标准为:校正p-value < 0.005 作为有显著性差异。 8.质谱成像(Air-flow assisted desorption electrospray ionization mass spectrometry imaging,AFADESI-MSI)分析肠癌及癌旁组织的代谢物:结直肠组织样本(包括腺瘤,肠癌以及癌旁组织)进行AFADESI-MSI空间代谢组学分析。 9.肠道微生物组-血清代谢组相关分析:使用血清和粪便匹配的33名结直肠癌患者进行肠道菌群与血清代谢物之间的相关性分析,相关性分析的方法为Pearson’s correlation。 10.选择肠道菌群相关代谢物用于肠癌检测模型的建立:LASSO algorithm 用于肠癌检测模型建立所需的肠道菌群相关代谢物的选择。
研究结果
1.非靶向代谢组学分析发现结直肠癌和腺瘤患者血清代谢物相较健康对照发生显著改变:为研究血清代谢组与结直肠腺瘤或肠癌之间的关系,发现队列样本进行非靶向代谢组学分析(图1.A)。发现队列分为三组,健康对照组(N, n = 31),腺瘤组(A, n = 12),肠癌组(C, n = 49)(图1.B)。滤除平均丰度< 5000 的低丰度信号,按比例混合的预期值和检测数值的线性回归模型R2 值见图1.C,50% 被检测代谢物的R2 值大于0.9,表明质谱检测的准确性。而且质控品批间的变异系数(coefficient of variances)小于15%。
不同组人群比较(C vs N、A vs N 和 C vs A,图1.D)后发现显著改变的代谢物,揭示了腺瘤和肠癌患者具有相似的代谢模式,而正常人群可以与这两种人群明显区分开来。进一步比较发现,C vs N 比较与 A vs N 比较中寻找到的特征代谢物有明显的相似性,表明在腺瘤阶段肿瘤就已经诱导了血清代谢组显著改变(图1.E)。这些显著改变的特征代谢物(共1426个)被命名为“结直肠异常相关代谢物”,其中885个可以被鉴定。这些代谢物的相对丰度见图1.F。基于这些代谢物,可以明确区分结直肠异常患者(肠癌C和腺瘤A组)和正常个体(图1.G)。
2.结直肠异常患者血清样本中的肠道菌群相关代谢物显著改变:这些改变主要体现在粪便肠道菌群与血清代谢组的整合分析、血清中特定胆汁酸代谢物的靶向分析、质谱成像技术揭示肠癌组织中菌群相关代谢物上调结果。
(1)粪便肠道菌群与血清代谢组的整合分析:为进一步研究肠道菌群与结直肠患者血清代谢物之间的相关性,并确定这些菌群相关代谢物对检测结直肠异常的潜在作用,在血清和粪便匹配队列(44个样本)进行了粪便宏基因组-外周血代谢组整合分析,获取血清样品的代谢组谱和粪便样品的宏基因组谱(图2.A)。从宏基因组数据中发现引起肠癌的关键病原体-产肠毒素脆弱拟杆菌 (ETBF) 丰度升高(图2.B,红色)。其他几种促进 肠癌的微生物均显著上调(图2.B,红色),而长双歧杆菌等益生菌在 肠癌患者中下调(图2.B,蓝色)。从宏基因组数据中筛选出相对丰度高于0.1%的640个菌种,在33例肠癌患者中检测到的这640个菌种与差异代谢物进行了Pearson相关系数分析(p value < 1E-3),而且此时的FDR (false discovery rate) 为 18% (图2.C)。发现322种结直肠癌异常相关代谢物与肠道菌群有显著关联,包括与肠癌发生和进展相关的菌种(图2.D)。通过评估这些代谢物预测结直肠癌和腺瘤的能力,发现其中63种代谢物可以解释87%的总变异。结直肠癌和腺瘤患者中促进肿瘤的菌群相关代谢物显著富集,而益生菌相关代谢物在正常人群中富集,根据代谢物分布可以明确区分正常个体和结直肠异常患者(图2.E)。
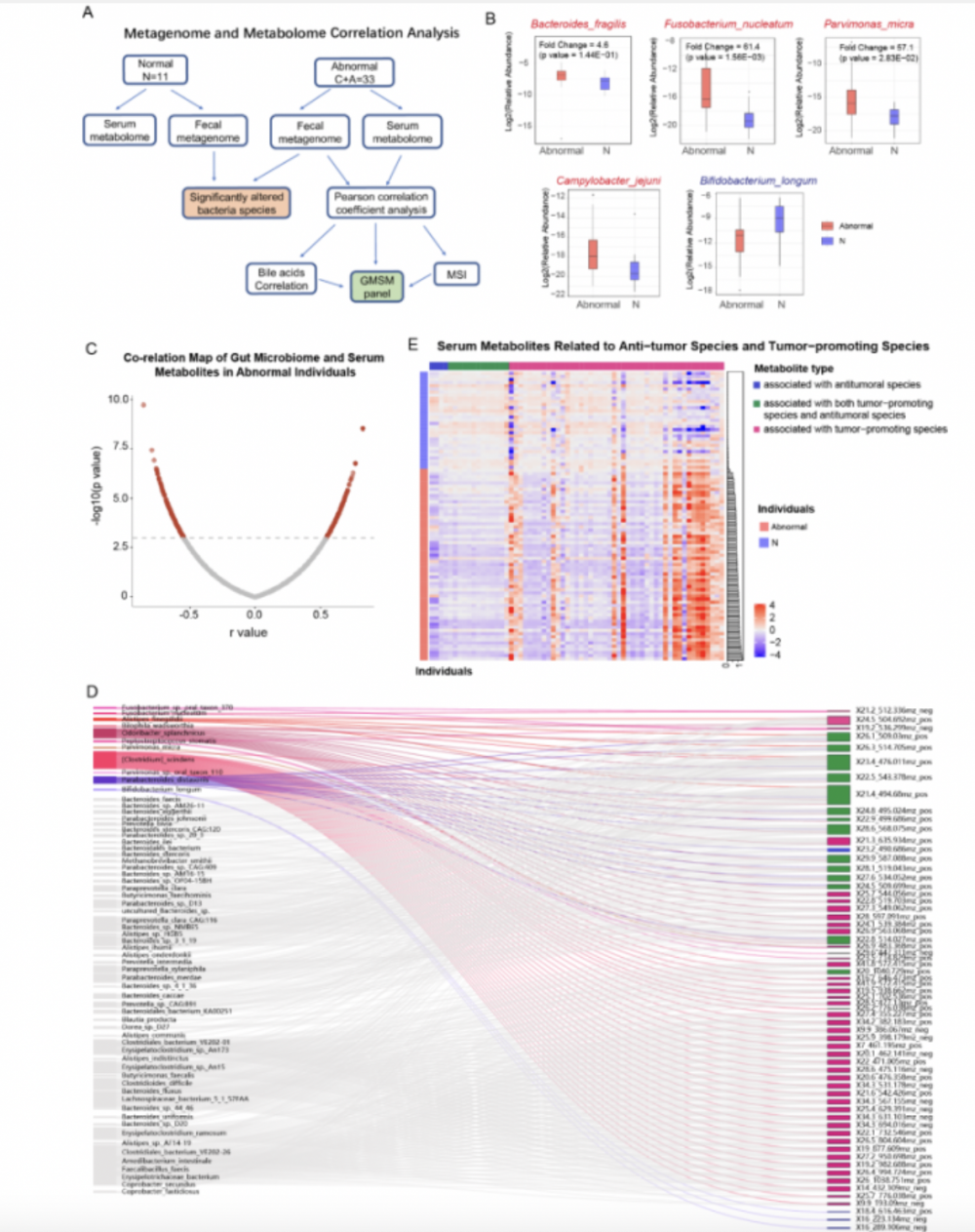
注:A:皮尔逊相关分析粪便样本和血清样本配对队列粪便宏基因组和血清代谢组用于获得肠道菌群相关代谢物。B肠癌相关的肠道菌群的相对丰度(红色:肿瘤促进菌种;蓝色:益生菌)。C:皮尔逊相关系数分布 (cut-off: p value < 1E-3, FDR ≤ 18%)。D:Sankey图示肠癌相关肠道菌和相关的血清代谢物。 E:肠癌促进菌种相关代谢物的丰度(紫色),抗肿瘤菌种相关代谢物的丰度(绿色),即抗肿瘤又促进肠癌的菌种相关代谢物(蓝色)。肠癌促进菌种相关代谢物在肠癌和腺瘤患者中显著富集。
图 2 肠道菌群相关代谢物在肠癌和腺瘤患者血清中显著改变
(2)血清中特定胆汁酸代谢物的靶向分析:为了进一步支撑上述发现的代谢物与肠道菌群之间的关系,次级胆汁酸代谢与肠道菌群和肠癌进展密切相关,进一步分析结直肠异常患者与正常个体的游离初级胆汁酸 (CA) 和脱氧胆汁酸 (DCA) 丰度的变化。在结直肠异常患者中,CA和DCA的血清浓度上调(图3. B, C),相关性分析揭示了肠道菌群与这些胆汁酸的关联(图3. D),而且这些相关的菌群在肠癌患者的粪便中其丰度上调。
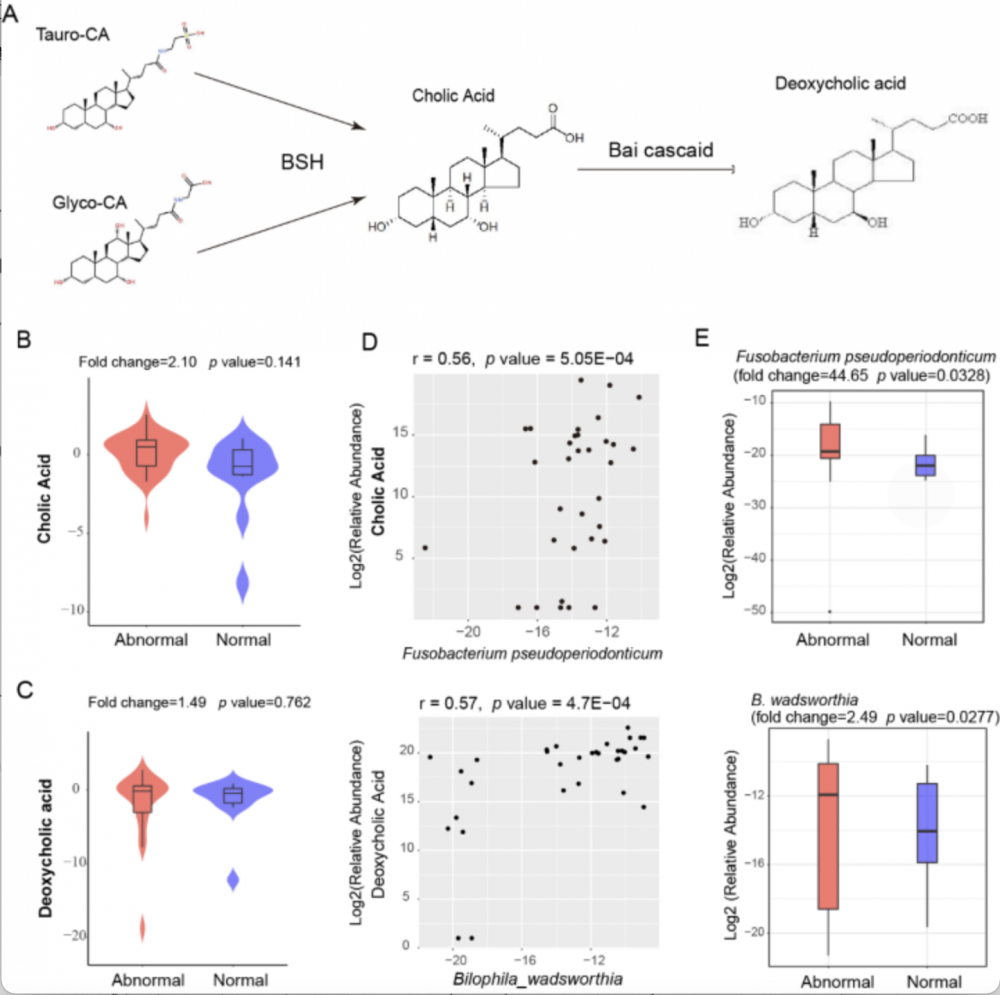
注:A. 次级胆汁酸在肠道菌群作用下的代谢途径。B. 非结合胆汁酸CA在肠道异常患者血清中上调。C. 非结合胆汁酸DCA在肠道异常患者血清中上调。D. 相关性分析表明非结合 CA 和 Fusobacterium pseudoperiodonticum,DCA 和Bilophila wadsworthia 正相关。E. 肠道菌群 Fusobacterium pseudoperiodonticum 和 Bilophila wadsworthia 在肠道异常患者粪便中富集。
图3 肠癌和腺瘤患者次级胆汁酸和相关菌群丰度改变
(3)质谱成像技术揭示肠癌组织中菌群相关代谢物上调:进一步使用质谱成像技术(AFADESI-MSI)检测了结直肠组织中的肠道菌群相关代谢物是否发生改变。9对结直肠腺瘤/肿瘤和癌旁活检组织,对其代谢物的相对含量进行检测(图4)。与邻近的正常组织相比,肿瘤/腺瘤组织中代谢物N,O-双-(三甲基甲硅烷基)苯丙氨酸的丰度显著上调(图4.A),在结直肠异常患者的血清中也观察到类似的变化(图4.B)。在33名结直肠异常患者中,该代谢物与Clostridiales bacterium VE202-01 和 Erysipelatoclostridium ramosum有显著正相关性,且这些微生物的相对丰度在结直肠异常患者中也上调(图4.C),这些实验结果表明肠癌相关菌群重塑可能对此代谢物的生物合成有正向调节作用,而该变化也在血清代谢组中反映出来。上述实验结果表明肠道菌群对外周血代谢物的构成及丰度产生影响。
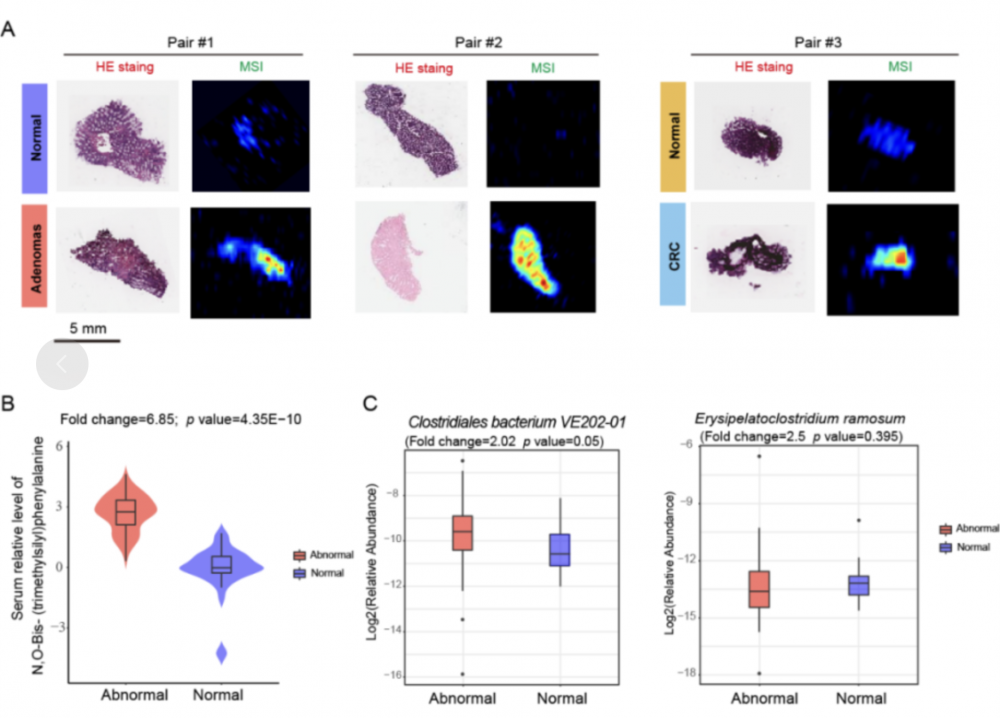
注:染色和MSI 表明 N,O-Bis-(trimethylsilyl)phenylalanine 在癌组织中上调 (A), 血清中的丰度也显著上调 (B). Clostridiales bacterium VE202-01 和 Erysipelatoclostridium ramosum在肠道异常患者血清中上调 (C)。
图4 HE (hematoxylin-eosin) 染色
3.肠道菌群相关代谢物(GMSM,gut microbiome-associated serum metabolites)能够发现结直肠异常患者;基于已经发现的322 个GMSM,使用 LASSO算法识别检测结直肠异常的关键代谢物(图5.A)。共计32个代谢物被筛选出来,其中8种代谢物能被准确定性,并在非靶向和靶向代谢组学检测中数据显示一致的上调或下调趋势,表明可以使用不同的方法准确测量这些代谢物(图5.A,B和Table.1)。 注:A:LASSO算法用于GMSM组合筛选。B:参考代谢物左: (Z)-5,8,11-trihydroxyoctadec-9- enoic acid (X14.3_329.233mz neg); 右: (E)-2-(4,8-dimethylnona-3,7-dien-1-yl)-5-hydroxy-2,7-dimethyl-2H-chromene-8-carbaldehyde (X27.8_353.212mz_neg).C:基于GMSM组合发现队列非靶向代谢组检测肠癌和腺瘤患者的ROC曲线。D:基于GMSM组合发现队列非靶向代谢组检测PCA分析。E:基于GMSM组合发现队列靶向代谢组检测肠癌和腺瘤患者的ROC曲线。F:基于GMSM组合发现队列靶向代谢组检测PCA分析。G:基于GMSM组合配对队列非靶向代谢组检测肠癌和腺瘤患者的ROC曲线。H:基于GMSM组合配对队列非靶向代谢组检测PCA分析。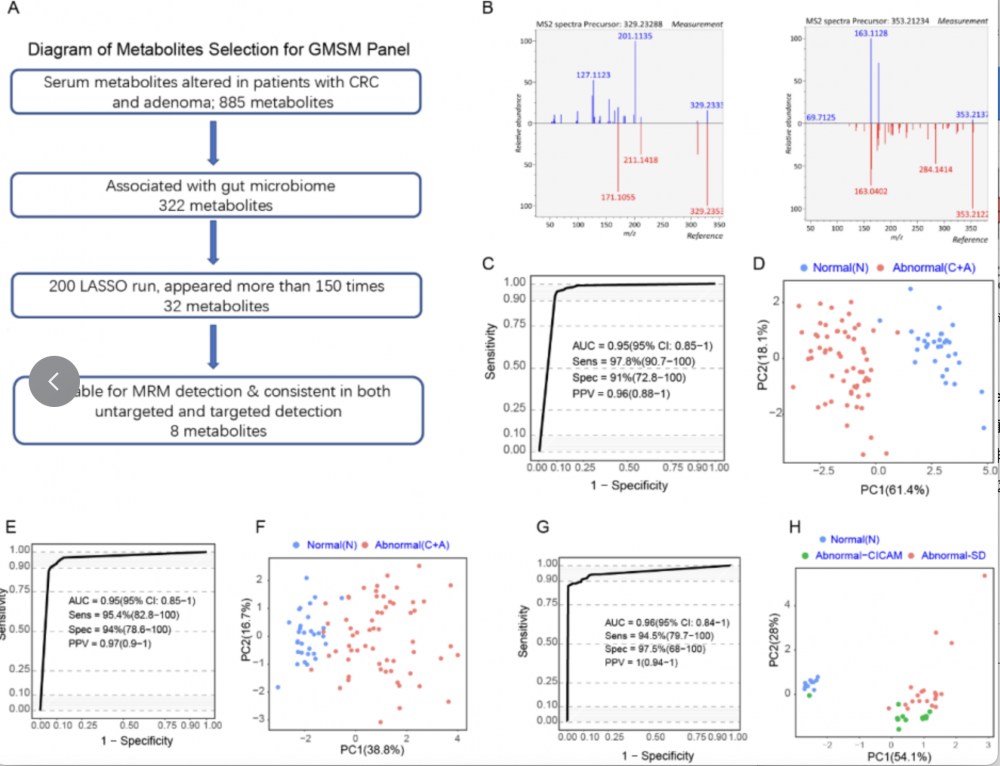
图 5 菌群相关血清代谢物可用于检测肠癌和腺瘤
表1 非靶向和靶向代谢组学发现的肠道菌群相关代谢物
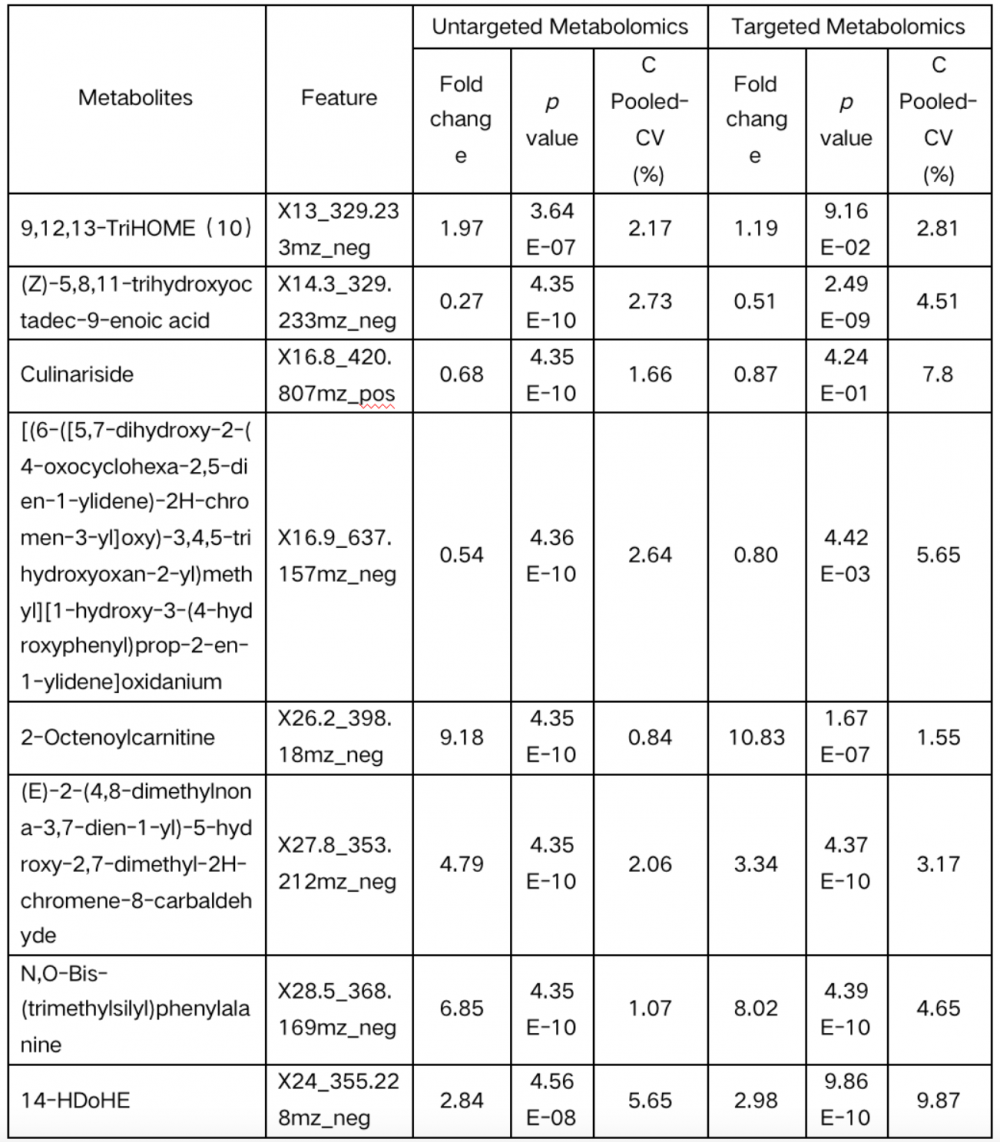
进一步评估此8个代谢物用于区分发现队列中的正常个体和结直肠异常患者的检测准确性。非靶向代谢组学分析结果可以准确区分发现队列中的正常个体和结直肠异常患者,曲线下面积 (AUC) 可达0.95(图5. C)。在靶向代谢组学中此8种代谢物的AUC为 0.95(图5.E),与非靶向代谢组学分析中获得的结果相似。PCA 图表明使用非靶向和靶向代谢组学分析,正常个体和结直肠异常患者之间存在明显区别(图5.D,F)。这8种代谢物在区分血清和粪便匹配队列中的腺瘤/肠癌与正常个体方面也表现出优异的准确性,AUC为 0.96(图5. G)。综上所述,此8个肠道菌群相关代谢物具有检测结直肠异常的潜力。
4.基于GMSM组合的预测模型在验证队列中检测腺瘤和肠癌患者的检测结果:根据发现队列中鉴定的代谢物,在建模队列中使用靶向方法测量8种GMSM组合的丰度,并用逻辑回归方法生成诊断模型,在建模队列用于肠癌和腺瘤诊断模型的AUC达到0.98(图6. A)。当cut-off值设为 0.438时(图6.B),建模队列中的敏感性为 96.7%,特异性为 90.3%。
进一步在验证队列中评估GMSM模型的诊断性能,其AUC达到0.92、灵敏度为83.5%、特异性为84.9%(图6. C)。该模型对于早期/中期(I/II期)肠癌患者的诊断其AUC为0.93;而对于晚期(III/IV期)其AUC达到0.91(图6.D)。对结直肠腺瘤和早期/中期 CRC(I/II 期)的敏感性分别达到 63.2% 和 88.2%,特异性84.9%。这些数据表明GMSM模型在早期检测肠癌方面的潜力。
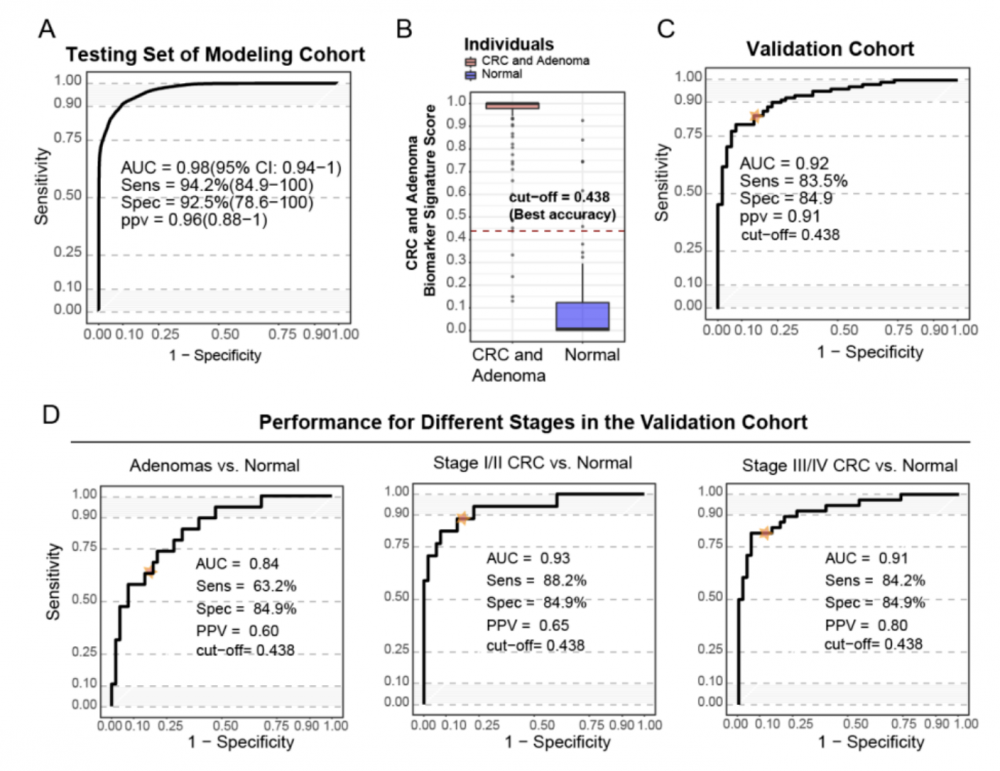
图 6 基于GMSM组合建立的诊断模型可以准确诊断肠癌和腺瘤患者
5.GMSM模型用于检测结直肠异常其效能优于目前临床使用的生物标志物CEA和FOBT:为了比较临床使用的标志物CEA和GMSM模型检测肠癌和腺瘤的效率,进一步评估了两种标志物在验证队列中的诊断效能。CEA的评估结果表明,其AUC为 0.72、敏感性为 35.8%、特异性为86.4%。相比之下,GMSM模型的AUC达到了0.92、灵敏度83.5%,特异性84.9%,远高于CEA(图7. A,B)。将GMSM模型与目前用于肠癌筛查的 FOBT/FIT分析进行了比较。共计89名接受过 FOBT/FIT检测的患者,其中58 名FOBT/FIT测试阳性,敏感性为 65.2%,与之前的报道水平相当。这些结果表明本研究建立的GMSM模型优于目前检测肠癌的FOBT/FIT检测。
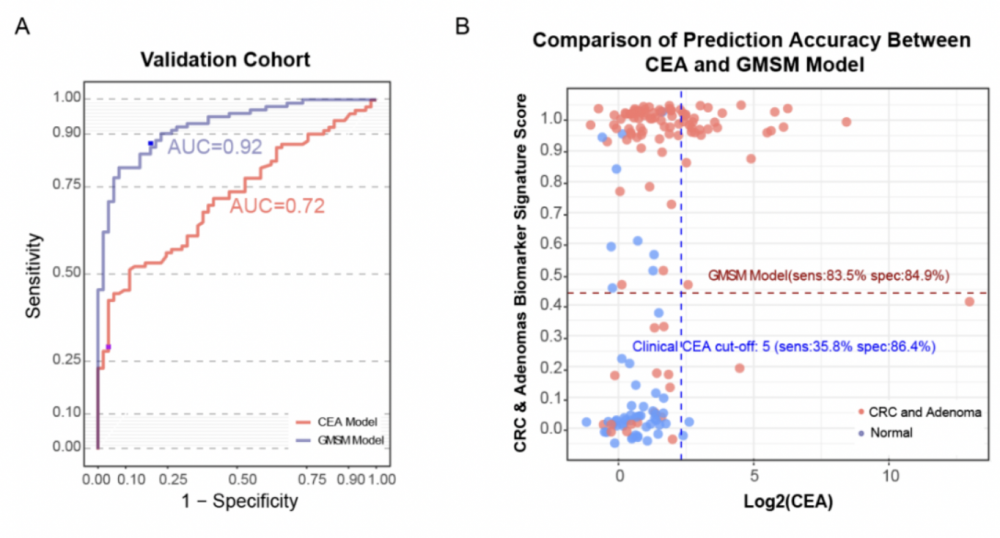
图 7 基于GMSM 组合建立的肠癌诊断模型的诊断效能与CEA的比较
讨论与分析
代谢组学检测日益成为肿瘤的检测手段,本研究利用代谢组学分析建立的肠癌和腺瘤检测模型,表现出比目前临床使用的生物标志物CEA和最近报道的蛋白质标志物及cfDNA突变的血浆生物标志物更高的准确性。GMSM组合是一种非侵入性的肠癌和腺瘤检测方法,与之前报道的代谢组(仅比较正常和肠癌个体之间的血清代谢组)结果相比,本研究的 GMSM 组合由一些特征代谢物组成,这些代谢物是肠癌相关的肠道菌群组改变的结果。
数学模型表明70-90%的癌症风险来自于环境,而肠道菌群是肠道最大的环境因素。最新证据表明,肠道菌群可能通过不同的机制诱导肿瘤发生和肠癌进展。本研究中发现肠癌患者的产肠毒素脆弱拟杆菌(ETBF, 图2.B)升高,脆弱拟杆菌毒素使结肠上皮细胞HT29/c1和T84中的精胺氧化酶(SMO,spermine oxidase)上调,导致产生SMO依赖的活性氧 (ROS,reactive oxygen species) 和诱导DNA损伤。此外细菌的几种代谢产物(例如次级胆汁酸)也与肠癌相关,结直肠异常患者的血清中游离胆汁酸(CA)和脱氧胆汁酸(DCA)浓度上调(图3.B)。相关性分析揭示了肠道菌群与胆汁酸之间的关联(图3.D),胆汁酸产生的ROS和活性氮物质导致DNA损伤,进而促进肠癌的发生。因此,本研究聚焦于肠道菌群相关代谢物获得的诊断模型用于肠癌的诊断灵敏度可达83.5%、特异性可达84.9%,高于其它血清标志物。最近发表的一项研究表明,基于粪便样本的11个微生物标志物,用于肠癌患者的检测,其AUC为0.80。而血液中的代谢物与各种生理或病理条件下的肠道菌群有关。因此通过粪便样本的代谢组学和宏基因组学整合分析,可以了解来自结直肠腺瘤和肠癌的特定肠道菌群和代谢物特征。
本研究通过相关性分析揭示了受肠道菌群影响的血液代谢物。首先确定了一系列与肠道微生物组(包括 F. nucleatum和B. longum菌,已经被证明促进或抑制肠癌生长)相关的代谢物。通过两项实验进一步评估本研究中肠道微生物组与血清代谢物之间的关联。对CA或DCA与肠道微生物组之间关联分析表明,CA和DCA的浓度与具有胆汁酸催化活性的细菌高度相关。肠癌患者血清和肿瘤组织中改变的特定代谢物(例如微生物代谢物 N,O-双-(三甲基甲硅烷基)苯丙氨酸)在血清和结直肠异常组织中均增加。这些代谢物与一些特定细菌之间的正相关性表明肠癌患者的肠道菌群重塑,菌群的变化可通过血清中代谢物的检测进行监测。通过正常人和肠癌人群粪便宏基因组和血清代谢组的整合分析,发现了结直肠异常与血清代谢特征之间的关联,据此建立的基于代谢物的预测模型,显示出用于准确检测肠癌的潜力。