


临床医生说:作为检验技师,能有这样的临床思维,难能可贵
单位:广州市花都区妇幼保健院(胡忠医院)检验科
电解质测定常用于判断体内是否有电解质紊乱情况,为临床疾病的诊疗提供依据,临床上,导致电解质紊乱的原因有很多。今天和大家分享一个笔者在工作中遇到的特殊的电解质紊乱案例,该患者多次高钾低钠,究其原因竟是一种罕见疾病——先天性肾上腺皮质增生症(Congenital Adrenal Cortical Hyperplasia,CAH)引起的。
患者系14天新生儿,性别不定,于2023年7月21日在我院剖宫产出生,出生后因“1、新生儿肺炎;2、三尖瓣反流(轻度);3、新生儿贫血;4、两性畸形?”,于7月21日收至我院新生儿科住院治疗,7月25日好转出院。出院后混合喂养,母乳为主,吃奶一般,具体不详,大便正常,小便2-3次/天。8月3日患儿出现吃奶减少,伴有呕吐黄色胃内容物、量少,无发热、咳嗽、气促、发绀,无抽搐等,为求进一步诊治,遂拟“吃奶少查因:新生儿感染?”于8月4日收入我院新生儿科。
T:36.5℃,P:140次/分,R:42次/分,BP:72/35mmHg,Wt: 2.42Kg。神志清,反应一般,肤色欠红润,皮肤弹性欠佳,轻度黄染,CRT2-3秒,唇无发绀,前囟平软,张力不高。呼吸欠规则,双肺呼吸音粗,未闻及明显干湿性啰音。心脏听诊未有明显异常,腹诊正常。原始反射可引出。外生殖器发育异常,酷似女性外阴,未见明显大小阴唇,未扪及睾丸。
1、先天性肾上腺皮质增生症;2、新生儿感染;3、高钾血症;4、低钠血症;5、氮质血症;6、新生儿贫血;7、凝血功能异常;8、新生儿高血糖。

即使是对于有着多年工作经验的笔者来说,像这样严重的高血钾、低血钠新生儿患者还是极少遇到。为了找出导致患儿高钾低钠的背后“元凶”,笔者仔细查阅患者的病历,并追踪查看一段时间内该患儿的结果,发现患儿多次出现危急值(高钾低钠),如图1、图2。
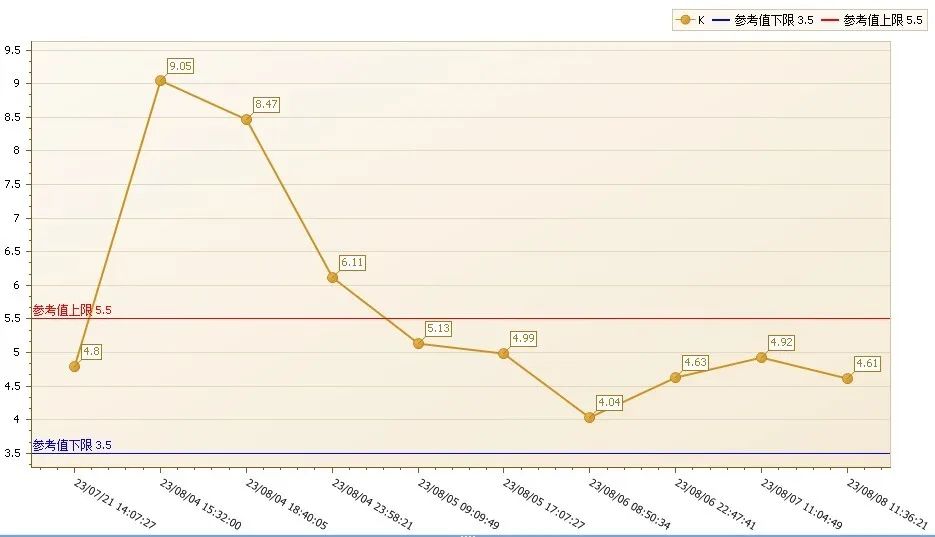
图1 血清钾的历史结果
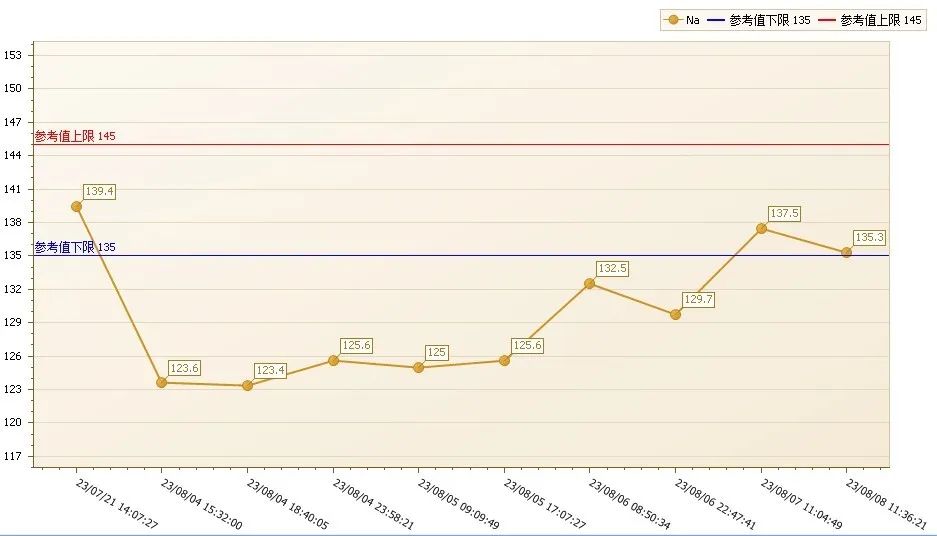
图2 血清钠的历史结果
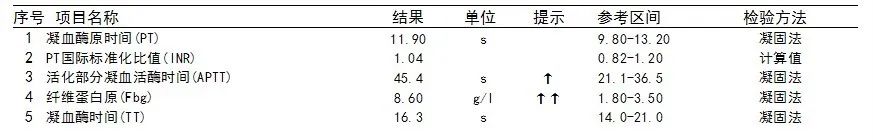
患儿的白细胞及PCT增高,其余未见异常,考虑存在感染;活化部分凝血活酶时间(APTT)升高和纤维蛋白原(Fbg)明显升高且达我院危急值范围,表明患儿凝血功能异常。
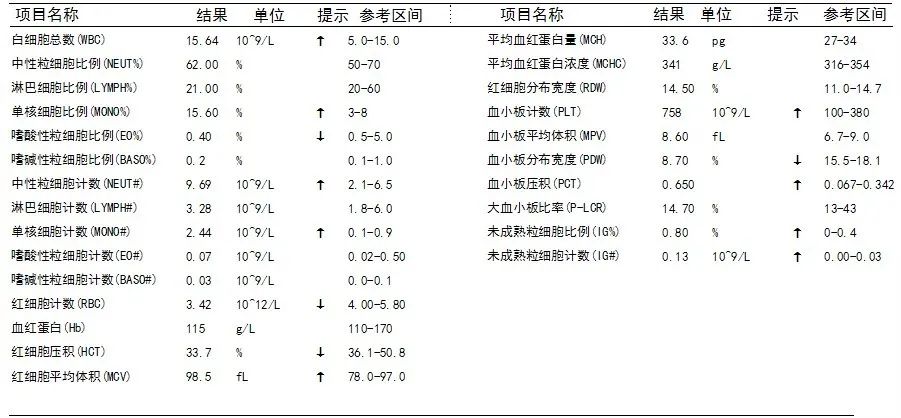

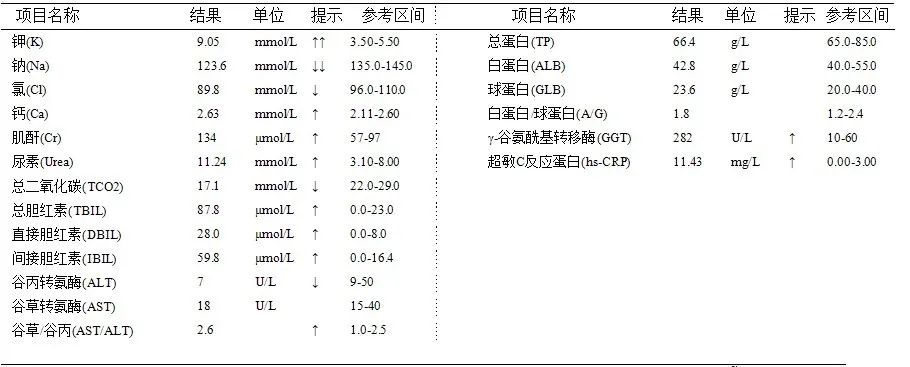
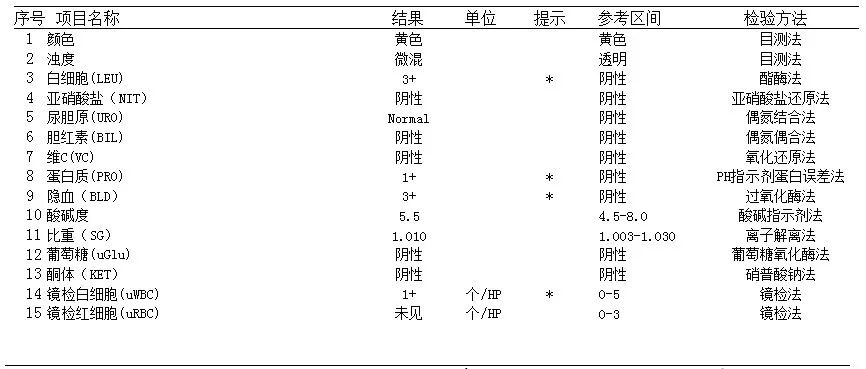
患儿的肌酐和尿素明显增高,考虑为氮质血症,尿常规多项结果异常,说明患儿已出现肾损伤。
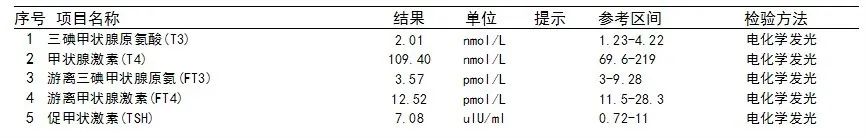
激素类项目甲功五项结果正常,睾酮(TESTO)增高,外送项目硫酸去氢表雄酮(DHEAS)、促肾上腺皮质激素(ACTH)、17α-羟基孕酮(17α-OHP)、醛固酮(ALD)均提示增高。
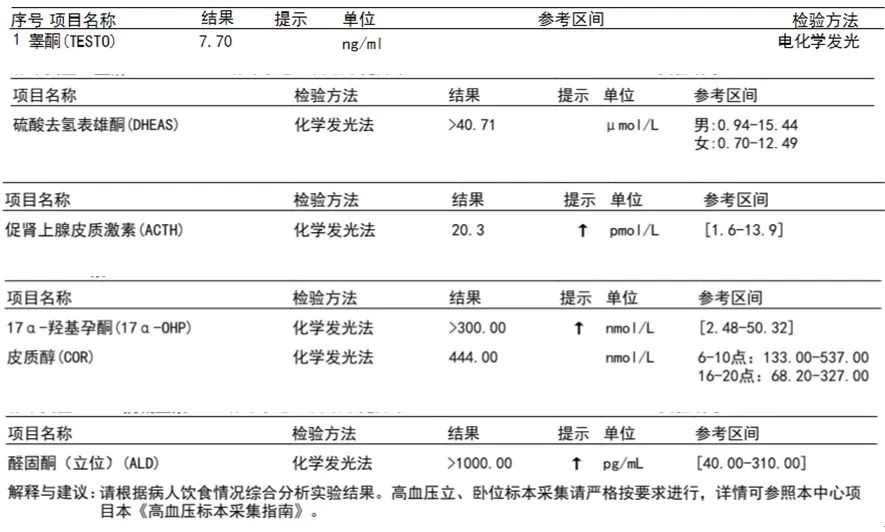
睾酮和硫酸去氢表雄酮属于雄激素,雄激素在男性主要是由睾丸和肾上腺分泌,在女性主要是由卵巢和肾上腺分泌。正常情况下,婴儿时期的性激素水平是很低的。案例中患儿生后外生殖器异常,酷似女性外阴,未见明显大小阴唇,未扪及睾丸,同时出现高雄激素,需高度怀疑先天性肾上腺皮质增生症(CAH)。
患儿的促肾上腺皮质激素升高,符合CAH的特点,CAH系由于肾上腺皮质激素合成中某些酶先天性缺陷,肾上腺皮质激素合成受阻,分泌不足,反馈性促进CRH及ACTH释放,所以CAH患者的ACTH升高。
患儿的血清17α-羟基孕酮结果明显升高(>300.00nmol/L),血清17-OHP 升高是21-羟化酶缺乏早期诊断的主要监测指标且具有较高的特异性,而CAH最多见的形式是21-羟化酶缺乏,21-羟化酶可催化17-羟孕酮转化为11-去氧皮质醇,该酶缺乏导致17-OHP堆积,故17-OHP明显升高是诊断CAH的可靠依据。
患儿的皮质醇不低、醛固酮水平明显升高,这不符合CAH的特点。有研究[1]表明诊断为CAH的患者只有23.26%会出现皮质醇减低,说明皮质醇辅助诊断CAH的特异性并不高。另有研究证明醛固酮水平对样本处在新生儿期及存在低血容量时参考价值不大[2]。患儿的醛固酮水平显著升高伴高钾和性发育异常,可排除原发性醛固酮增多症,怀疑是高钾血症导致的醛固酮应激性增高。
CAH确诊的金标准是基因检测。对于临床疑似而生化诊断困难者,或诊断不明但已用糖皮质激素治疗者,通过基因分析有助确诊。CAH患者,需先进行染色体核型检查,明确其性染色体,再进行基因检测。
综合分析,患儿生后发现外生殖器异常,查17-羟基孕酮明显升高,生化指标提示高钾血症、低钠血症,考虑先天性肾上腺皮质增生症,21-羟化酶缺陷(21-OHD),分型方面为典型失盐型可能性大,临床治疗方面需用氢化可的松及氟氢可的松治疗。
CAH按照酶缺陷类型的不同,可分为21-羟化酶缺乏症、11β-羟化酶缺乏症、3β-羟类固醇脱氢酶缺乏症、17α-羟化酶缺乏症、20,22-碳链酶缺乏症、类固醇激素急性调节蛋白缺陷症及17β-羟类固醇脱氢酶缺陷症等,最常见的是21-羟化酶缺陷(21-OHD),占 90%~95%[4],其致病原因是CYP21A2基因突变引起的。
本案例患儿从入院病因不明到最后诊断为先天性肾上腺皮质增生症,充分体现了实验室检查在临床诊疗中的重要性。同时在日常工作中,检验人员遇到异常结果或危急值结果时,应积极与临床沟通,协助临床医生更快的找到患者的具体病因,从而给予患者更快更有效的治疗。

参考文献
[1]刘西芳,戴立英等. 新生儿先天性肾上腺皮质增生症的诊断[J]. 山东医药,2022,62(36),57-59.
[2]周莛,朱岷. 儿童肾上腺危象临床特征及基因分析[J]. 医学信息,2020,33(9):67-72.
[3]El-Maouche D,Arlt W,Merke DP. Congenital adrenal hyperplasia[J].Lancet,2017,390(10108):2194-2210.
[4]Merke DP,Auchus RJ. Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency[J]. The New England journal of medicine,2020,383(13):1248-1261.
内容来源:医学检验沙龙
图片来源:veer、ibaotu
排版:znm
审校:金宝


