


神经系统自身免疫病相关自身抗体谱检测技术与临床诊疗思路
 黄晶,吉林大学第一医院检验科主任,教授、主任医师、博士研究生导师。中国检验检疫学会卫生检验与检疫专业技术委员会副主任委员、中华医学会检验分会临床免疫学组委员、中国医师协会检验分会自身免疫专业委员、中国中西医结合学会检验医学专业委员会常委、吉林省中西医结合学会检验专业委员会副主任委员、吉林省医学会检验分会常委、吉林省检验质量控制中心副主任、长春市医学会检验分会主任委员,中华检验杂志审稿专家等。从事实验诊断学临床感染免疫、自身免疫与肿瘤免疫及临床免疫学项目的实验室检测、临床应用及相关体外诊断试剂的研究。主持/参与国家级、省部级、市级、院校级课题30余项。发表论文90余篇,其中SCI论文9篇(总IF29.2),获得省科技进步二等、三等奖及吉林大学医疗成果、教学成果奖多项,出版专著1本。
黄晶,吉林大学第一医院检验科主任,教授、主任医师、博士研究生导师。中国检验检疫学会卫生检验与检疫专业技术委员会副主任委员、中华医学会检验分会临床免疫学组委员、中国医师协会检验分会自身免疫专业委员、中国中西医结合学会检验医学专业委员会常委、吉林省中西医结合学会检验专业委员会副主任委员、吉林省医学会检验分会常委、吉林省检验质量控制中心副主任、长春市医学会检验分会主任委员,中华检验杂志审稿专家等。从事实验诊断学临床感染免疫、自身免疫与肿瘤免疫及临床免疫学项目的实验室检测、临床应用及相关体外诊断试剂的研究。主持/参与国家级、省部级、市级、院校级课题30余项。发表论文90余篇,其中SCI论文9篇(总IF29.2),获得省科技进步二等、三等奖及吉林大学医疗成果、教学成果奖多项,出版专著1本。
 车媛媛,吉林大学第一医院检验科副主任医师,临床免疫学博士,研究方向:自身免疫病相关抗体的实验室检测。主持国自然青年基金1项,第一作者发表SCI文章4篇(总IF20.553)。
车媛媛,吉林大学第一医院检验科副主任医师,临床免疫学博士,研究方向:自身免疫病相关抗体的实验室检测。主持国自然青年基金1项,第一作者发表SCI文章4篇(总IF20.553)。
一、自身免疫性脑炎与相关抗体
2017版《中国自身免疫性脑炎诊治专家共识》对自身免疫性脑炎(autoimmune encephalitis,AE)的定义是泛指一类由自身免疫机制介导的脑炎。根据免疫学发病机制分为针对神经元胞内抗原成分,介导细胞免疫而引起不可逆神经元损害的经典副肿瘤性边缘性脑炎,和靶抗原分布于神经元表面或突触蛋白,介导体液免疫引起相对可逆神经元功能障碍的一类新型AE。后者又根据各自代表性抗神经元抗体及对应的不同临床表现分为抗N-甲基-D-天冬氨酸受体(NMDAR)脑炎、边缘性脑炎和其他AE综合征[1]。
1. 抗NMDAR脑炎:抗NMDAR脑炎最常见,临床表现也最为多样,以抗NMDAR抗体阳性和弥漫性脑炎为特征性表现,约占AE患者的80%,患者可以主要表现为精神行为异常、认知障碍、近记忆力下降、癫痫发作、言语障碍、运动障碍、不自主运动、意识水平下降与昏迷、自主神经功能障碍等,也可部分出现睡眠障碍、中枢神经系统局灶性损害所致的复视、共济失调和肢体瘫痪以及周围神经和神经肌肉接头受累引起的神经肌强直、肌无力或腹泻等。NMDAR抗体相关脑炎确诊标准强调症状与脑脊液抗NMDAR抗体阳性两个要素[1,2]。
2. 边缘性脑炎:边缘性脑炎以精神行为异常、癫痫发作和近记忆力障碍为主要症状,抗富含亮氨酸胶质瘤失活蛋白1(LGI1)抗体相关脑炎、抗γ氨基丁酸B型受体(GABABR)抗体相关脑炎和抗α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体(AMAPR)抗体相关脑炎属于这一类,发病率低于NMDAR脑炎。边缘性脑炎对应的抗体种类较多,不同类型抗体阳性对应的临床表现多样,临床研究发现,LGI1和CASPR2抗体阳性可以作为难治性癫痫的辅助诊断指标,且与CASPR2抗体患者(53%)相比LGI1抗体阳性患者更常发生癫痫(90%),且以局灶性发作性癫痫为主。2岁以下儿童的高滴度CASPR2抗体阳性与Morvan综合征有关,而高血清滴度的GAD抗体与病因不明的成人发作性颞叶癫痫(TLE)有关,GABABR抗体与伴有严重癫痫发作的边缘叶脑炎的进展有关,且合并肿瘤多发生于男性患者中[3-6]。
3. 其他AE综合征:包括Morvan综合征、抗GABAAR抗体相关脑炎、伴有强直和肌阵挛的脑脊髓炎(PERM)、抗二肽基肽酶样蛋白(DPPX)抗体相关脑炎、抗多巴胺2型受体(D2R)抗体相关基底节脑炎以及抗IgLON5抗体相关脑病等,这些AE综合征可同时累及中枢神经系统和周围神经系统,也可以表现为特征性的临床综合征,抗神经元抗体阳性是诊断的主要依据之一[1]。
4. 自身免疫性小脑炎:近年来,自身免疫性小脑炎(autoimmune cerebellitis,AC)又称自身免疫性小脑共济失调(autoimmune cerebellar ataxia,ACA),也越来越多的被研究者们关注,自身免疫性小脑共济失调涵盖的相关抗体更为多样。2015年Jarius等人发表名为"美杜莎共济失调"(Medusa-head ataxia)的综述[7],详细阐述了AC相关抗体多数在间接免疫荧光实验中与小脑浦肯野细胞的胞质和树突结合,包括GAD65、ZIC4、Tr/DNER、ITPR1、Homer-3、NCDN、CARP Ⅷ(Carbonic anhydrase-related protein Ⅷ,碳酸苷酶相关蛋白Ⅷ)和PCA2抗体等。2020年Ram N Narayan等将影响前庭终末器官、前庭通路、前庭核和前庭小脑的一类以慢性眩晕和共济失调为主要表现的神经自免病统称为自身免疫性前庭小脑综合征。明确神经元胞内抗原特异性抗体Yo(PCA-1)、Hu(ANA-1)、Ri(ANA-2)、Ma1/2(KLHL11/12)抗体、Amphiphysin、CV2(CRMP5)、AP3B2、PCA-2(MAP1B),NCDN,以及靶向神经元表面或突触抗原的特异性抗体Tr/DNER、CASPR2、septin-5、Homer-3和mGluR1、TRIM 46, 9 & 67、NIF、Septin 5、mGluR2、SEZ6L2[8、9]存在于自身免疫前庭小脑综合征患者中。强调了这些神经元表面、突触和胞内抗原特异性自身抗体可作为AC早期诊断的生物标志物。
表1. 自身免疫性脑炎相关自身抗体

二、 神经系统副肿瘤综合征与相关抗体
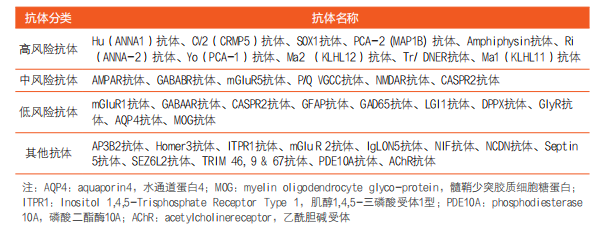
神经系统副肿瘤综合征(paraneoplastic neurologic syndrome,PNS)是发生在某些肿瘤或潜在肿瘤患者体内,肿瘤触发免疫应答反应,引起远程效应,对神经系统造成急性或亚急性损伤而出现的一系列神经综合征,在肿瘤患者中发病率约为300分之一。PNS损伤可累及中枢神经、周围神经、神经肌肉接头和肌肉等多个部位,出现相应的临床表现。随着对该病临床、免疫学、肿瘤异质性等方面研究的深入,新的表型和抗神经元抗体谱不断扩展,2021年美国神经病学学会提出了PNS更新版诊断标准。建议用“高危表型”取代“经典综合征”,7类高危表型包括脑脊髓炎(EM)、边缘性脑炎(LE)、快速进展性小脑综合征、眼肌阵挛综合征(OMS)、感觉神经病变(SNN)、胃肠道假性梗阻(肠源性神经病)和兰伯特-伊顿肌无力综合征(LEMS),代表性自身抗体有:Hu、CV2、LGI1、CASPR2、GABABR、AMPAR、Ma2、Yo、Tr/ DNER、Ri、Amphiphysin、AChR、VGCC、SOX1抗体。同时提出了“中间风险表型”的概念,将除边缘性脑炎以外的其他类型脑炎归为此类。旧版指南中的“神经肿瘤抗体”被“高风险”(与肿瘤相关性>70%)抗体取代。根据抗体在肿瘤中的出现频率,分为高风险、中风险(与肿瘤相关性30%-70%)以及低风险(与肿瘤相关性<30%)三个等级。同时,综合临床表型以及抗体与癌症之间的对应性将PNS诊断也分为三个等级:确定、很可能和可能。除眼肌阵挛外,强调确诊PNS需要存在高风险或中等风险抗体[10]。
新版指南还将一些新的神经系统疾病的检测抗体谱纳入PNS诊断标准中,包括:炎症性肌病(多发性肌炎和坏死性肌病)、重症肌无力、与单克隆抗体相关的多发性神经病、丙种球蛋白病、副肿瘤性视网膜病、视神经炎和耳蜗前庭病等。
随着研究的不断深入,一些新的PNS诊断指标也在陆续出现,如:PDE10A、AP3B2、ITPR1等[11、12]。这些指标的发现及其作用机制的探讨,必将推动PNS诊断标准的进一步更新完善。
表2. PNS相关自身抗体
三、中枢神经系统炎性脱髓鞘疾病与相关抗体
中枢神经系统(central nervous system,CNS)脱髓鞘疾病是一组脑和脊髓以髓鞘脱失影响神经元信号传导,从而导致运动、视觉和感觉障碍为主要特征的疾病,包括遗传性及获得性两大类。前者主要由遗传因素导致,统称为脑白质营养不良;获得性CNS脱髓鞘又分为继发性脱髓鞘(如缺氧缺血性脑病、营养缺乏性疾病)和原发性免疫介导的炎性脱髓鞘病。临床研究发现,CNS炎性脱髓鞘发病机制与自身抗体有关[13]。
1. 视神经脊髓炎(neuromyelitis optica,NMO):NMO是视神经与脊髓同时或相继受累的反复发作的CNS炎性脱髓鞘病变。部分患者可有脑部受累,出现脑病、惊厥等症状,还可累及CNS其他部位,表现为难治性呕吐或呃逆(延髓颈髓交界区受累)、日间过度嗜睡或发作性睡病(间脑)、可逆性后部白质脑病综合征及神经内分泌失调等。NMO特异性抗体的靶点是中枢神经系统血-脑屏障星形胶质细胞足突的水通道蛋白4(AQP4)[14],AQP4抗体滴度与NMO疾病活动度相关,且在NMO的发病机制中起着重要作用,AQP4抗体阳性被作为NMO的诊断标准之一,并用于NMO与多发性硬化(multiple sclerosis,MS)的鉴别诊断[15]。
2. 急性播散性脑脊髓炎(acute disseminated encephalomyelitis,ADEM):ADEM是一种广泛累及CNS白质的急性炎症性脱髓鞘病。多发于儿童和青年,病毒感染或疫苗接种或可诱发。临床研究发现,尽管抗髓鞘少突胶质细胞糖蛋白(MOG)抗体在孤立综合征、多发性硬化(MS)以及其他神经系统疾病中有表达,但高滴度的MOG更常见于年轻的ADEM患者中,且MOG抗体滴度下降往往提示ADEM预后良好[16],因此MOG抗体在ADEM的诊断和预后评估中具有参考价值。
3. 多发性硬化(multiple sclerrosis,MS):MS是一种以中枢神经系统白质炎症性脱髓鞘为主要病理特点的自身免疫病,发病机制不明。已证实MS患者中存在多种自身抗体,包括抗髓鞘抗体、抗神经元和轴突抗体以及其它自身抗体。(1)抗髓鞘抗体:抗髓鞘抗体分为抗髓鞘蛋白抗体和抗髓鞘脂质抗体,前者包括髓鞘碱性蛋白(MBP)抗体、髓鞘少突胶质细胞糖蛋白(MOG)抗体、髓鞘蛋白脂蛋白(PLP)抗体和髓鞘相关糖蛋白(MAG)抗体,其中抗髓鞘脂质抗体的主要类型为半乳糖脑苷脂(GalC)抗体。研究发现,MBP抗体在MS急性期、复发缓解期和进展期的血清检出率可达40%~70%[17、18],由MBP抗体介导的蛋白水解活性与MS患者扩展残疾状态量表(EDSS)评分相关[19],但其诊断特异性不高。MOG抗体在MS患者血清和脑脊液中均可检出,阳性率约为40%[20],与抗体阴性的MS患者相比,MOG和MBP抗体阳性患者的缓解期短、复发率高[21]。GalC抗体在MS患者中的阳性率约为23%,以复发-缓解型多发性硬化(RRMS)多见,且具有极高的特异性,因此,MOG、MBP和GalC抗体都可作为判断临床孤立综合征进展为MS的可靠指标[22],另外,抗髓鞘质抗体还是MS病情监控和神经变性的生物标志物[23]。(2)抗神经元和轴突抗体:抗神经元和轴突抗体与MS病情进展相关。神经微丝蛋白(Neuronal Filament,NF)可作为轴突损伤的生物学标志物。一项对自身抗体相关中枢神经系统疾病的临床观察显示,在临床孤立综合征、复发-缓解型多发性硬化、继发进展型多发性硬化(SPMS)和中枢神经系统非炎症性或其他炎症性疾病患者中,临床孤立综合征尤其是最终进展为MS的患者,中枢神经系统神经微丝蛋白轻链抗体滴度显著升高;而SPMS患者脑脊液神经微丝蛋白重链抗体滴度明显升高。此外在MS患者脑脊液和(或)血清中还发现了针对微管蛋白、神经节苷脂、Nogo受体、磷酸丙糖异构酶(TPI)等抗原的抗体[24-30]。(3)其他自身抗体:研究发现,MS患者脑脊液和血清中存在Nogo-A、NG2、SWAP70、热休克蛋白60和90,微管蛋白、神经节苷脂、磷酸丙糖异构酶(TPI)、抗亚硝基半胱氨酸IgM等抗体,其中Nogo-A和NG2与髓鞘再生和修复相关,SWAP70主要存在于复发-缓解型MS患者血清中,复发期MS脑脊液内抗亚硝基半胱氨酸IgM滴度较缓解期高[27-37]。
四、重症肌无力与相关抗体
2020版《中国重症肌无力诊断和治疗指南》对重症肌无力(Myasthenia Gravis,MG)的概念进行了明确描述。MG是由自身抗体介导的获得性神经-肌肉接头(neuromuscular junction,NMJ)传递障碍的自身免疫性疾病[38]。目前,已证实了MG患者血清中可能存在多种致病性抗体。指南推荐的MG亚组分类正是基于血清抗体和临床特征,证明血清学抗体的检测对MG精准治疗和预后评估具有指导意义。
1. 乙酰胆碱受体(acetylcholine receptors,AChR)抗体:AChR抗体是最常见的致病性抗体。对应的靶抗原为分布于NMJ突触后膜、部分前膜、神经节细胞突触表面以及胸腺肌样上皮细胞表面的一种跨膜糖蛋白。AChR抗体主要的致病亚型为IgG1、IgG3两型。通过抑制乙酰胆碱与受体结合的方式阻碍骨骼肌间神经信号传导。常以眼肌无力为首发症状(85%),后可累及全身。AChR抗体阳性对应的MG亚组包括:眼肌型MG(ocular MG,OMG)、AChR-全身型(generalized MG,GMG)(早发型)、AChR-GMG(晚发型)及胸腺瘤相关MG[38]。AChR抗体阴性的MG定义为血清学阴性MG(serological negative MG,SNMG)[39]。
2. 肌肉特异性酪氨酸激酶(muscle-specific tyrosine kinase,MuSK)抗体:MuSK抗体在MG患者血清中检出率约为1%-4%,对应的靶抗原是突触后蛋白MuSK,后者磷酸化能够引发AchR聚集,维持NMJ正常信号传导。MuSK抗体的主要致病亚型为IgG4,常存在于SNMG患者血清中,对应的MG亚组为MuSK-MG,以球麻痹、面颈肌无力为主要临床表现。
3. 低密度脂蛋白受体相关蛋白(LDL receptor related protein 4,Lrp4)抗体:在1%-5%的MG,7%-33%的AChR、MuSK阴性MG患者血清中可检出Lrp4抗体。靶抗原Lrp4活化后,能够结合MuSK,启动后者的磷酸化过程,参与信号传导。Lrp4抗体的主要致病亚型为IgG1,对应的MG亚组为Lrp4-MG,临床特点尚不明确。
4. 横纹肌抗体:有证据显示MG患者血清中还存在一类横纹肌抗体,包括连接素(Titin)抗体和兰尼碱受体(ryanodine receptor,RyR)抗体,Titin和RyR抗体对应的MG亚组有AChR-GMG(晚发型)和胸腺瘤相关MG,常伴随AChR抗体同时出现。这些抗体具体的作用机制和临床表型有待进一步研究[38,40]。
5. 其他抗体:此外,针对Agrin、胶原Q(collagen Q,ColQ)、Kv1.4钾离子通道、rapsyn、肌动蛋白、乙酰胆碱酯酶(acetylcholinesterase,AChE)和胶原XIII的自身抗体也在MG患者中被发现,这些抗体的病理机制和临床诊断价值有待进一步研究[40]。
五、免疫介导周围神经病与相关抗体
临床上免疫介导周围神经病以吉兰-巴雷综合征(Guillain-Barré syndrome,GBS)和慢性炎性脱髓鞘性多发性神经根神经病(chronic inflammatory demyelinating polyradiculoneuropathy,CIDP)最为常见。
1. GBS:2019版《中国吉兰-巴雷综合征诊治指南》中定义GBS系一类免疫介导的急性炎性周围神经病,以多发神经根及周围神经损害为主要临床表现[41]。GBS的亚型分类包括:急性炎性脱髓鞘性多发神经根神经病(acute inflammatory demyelinating polyneuropathies,AIDP)和急性运动轴索性神经病(acute motor axonal neuropathy,AMAN)、急性运动感觉轴索性神经病(acute motor-sensory axonal neuropathy,AMSAN)、Miller-Fisher综合征(MFS)和急性泛自主神经病和急性感觉神经病等。有证据表明发病机制与神经节苷脂自身抗体有关的是AMAN、AMSAN和MFS三型。最近的一项临床研究显示神经节苷脂抗体在GBS患者血清中检出率为61%,至少包含GM1,GM2,GM3,GD1b,GD3,aGM1,GT1a,GT1b和GQ1b抗体中的一项阳性,且最多见的是GM1,GD1b和GQ1b型。特异性抗体对临床表型具有指向性,且与预后有关。其中,GM1和GD1a抗体阳性常见于AMAN和AMSAN两型,GQ1b抗体IgG亚型是MFS的诊断标志物之一,GD1b抗体与急性共济失调性神经病有关,GT1a抗体可与GQ1b发生交叉反应,阳性多见于咽颈臂变异型,靶向GM1或GD1a的神经节苷脂IgG型抗体阳性与GBS预后不良相关,或可成为长期轴突损伤的标志物。另外,抗神经节苷脂复合物抗体也被发现可作为GBS诊断和预后评估的指标,如抗GD1a/GD1b复合物抗体与严重GBS有关,抗GM1/GalNAc-GD1a复合物抗体被发现与具有频繁传导阻滞的纯运动型GBS有关[42-48]。
2. CIDP:CIDP是一类由免疫介导的慢性进展或复发性运动感觉周围神经病。包括经典型和变异型两类,后者又分为纯运动型(pure motor CIDP)、纯感觉型(pure sensory CIDP)、远端获得性脱髓鞘性对称性神经病(distal acquired demyelinating symmetric,DADS)、多灶性获得性脱髓鞘性感觉运动神经病(multifocal acquired demyelinating sensory and motor neuropathy,MADSAM或Lewis-Sumner综合征)、局灶型(focal CIDP)等。抗郎飞结/结旁蛋白抗体被证实与多种CIDP的临床表型有关[49]。郎飞结区的离子通道是神经信号传导的重要通路,多种蛋白和细胞黏附分子在其间发挥作用。研究发现,5.5%的CIDP患者血清中存在针对结区和结旁区蛋白的自身抗体[50],靶抗原包括神经束蛋白155(neurofascin 155,NF155)、接触蛋白1(contactin-1,CNTN1)、接触蛋白相关蛋白1(contactin-associated protein1,CASPR1)等。一项大样本研究报道,NF155 IgG4型抗体在CIDP患者中检出率为7%,NF155 IgG4阳性患者以青年男性多见,表现为经典型CIDP时多出现肢体震颤症状,另外,以下肢远端无力及感觉障碍起病,并出现足下垂和步态异常时,临床表现符合DADS亚型。早期出现轴索损害常提示预后不良。8%的NF155 IgG4阳性CIDP患者合并CNS脱髓鞘改变。CNTN-1 IgG4型抗体在CIDP患者中检出率为2.4%,CNTN1 IgG4阳性CIDP好发于老年男性,且病程进展快,以运动系统受累为主,未见合并CNS脱髓鞘改变的报道。不同地域患病人群的感觉性共济失调存在较大临床异质性,可能与遗传因素有关。CASPR1 IgG4型抗体阳性CIDP多为急性或亚急性起病,表现为严重的疼痛和运动系统受累。最近的研究发现,伴有震颤的严重CIDP亚型与这些针对结区和结旁区蛋白的IgG4型致病性抗体有关[51]。
六、神经系统自身抗体的检测现状
神经系统自身抗体的检测方法主要有间接免疫荧光法、免疫斑点法和酶联免疫吸附法三种。间接免疫荧光法采用组织切片或重组细胞抗原作为检测基质,检测结果具有高灵敏度;免疫斑点法是将多种纯化抗原包被在固相膜载体表面,检测特异性强,且可实现多指标联合检测;酶联免疫吸附法主要用于特异性抗体的半定量/定量检测。随着检测技术的进步,多种方法学、多指标联合检测策略得以有效应用,其检测结果较单一指标能够提供更全面的信息,最大程度防止漏检,进而更快速、准确的辅助疾病诊断并早期个性化治疗。
目前,绝大多数的神经系统自身抗体都有相应的商品化检测试剂,并且在西方很多国家被有效应用于临床检测。但是,国内开展神经系统自身抗体检测的医院极为有限,检测项目也较局限。主要原因是:
① 国产试剂品种少,许多进口的商品化检测试剂盒尚未通过国家食品药品管理局的注册批准;
② 全国范围内缺乏针对神经系统自身抗体检测指标的收费标准;
③ 国内对于神经系统自身抗体认识不足,发病机制研究滞后;
④ 缺乏自身抗体检测新项目快速开展的准入机制;
⑤ 国内目前没有针对该类检测的室间质评项目,实验室间结果缺乏参比性。
为了解决上述问题,我们认为:
① 试剂生产厂家、政府相关审批部门应共同努力,加快试剂批号报批、物价审批速度,引入新项目快速开展的准入机制;
② 加强实验室与临床的沟通,根据临床需求开展相应自身抗体检测;
③ 制定相应的抗体检测技术标准与规范,有条件时应建立参考实验室,进行全面的质量控制与管理,使神经系统自身免疫病领域的临床数据可以相互参照。


