


宏基因组高通量测序技术临床转化与平台建设

李永军,微远基因首席执行官,中华预防医学会诊断微生态学会委员,北京医学检验学会微生物与感染分会常委,海峡两岸医药卫生协会精准感染专委会常委,广东省精准医学应用学会肝病分会常委;2006年-2010年工作于中国医学科学院病原生物学研究所,在EID、JCM、ERJ等杂志发表文章多篇。

苟雪静,中山大学微生物学硕士研究生,微远基因产品经理,广东省精准医学应用学会微生态医学分会委员,5年mNGS研发与临床应用经验,发表SCI文章5篇以上,影响因子20+,撰写mNGS解读与进展文章200余篇。
在临床感染病原学诊断过程中,时常会面对一些不明原因感染或受限于检测技术灵敏度无法获得准确结果的困境。临床迫切需要突破性的病原体诊断方法,直接、快速、准确的从临床样本鉴定感染病原体,进而指导临床治疗。随着测序技术的飞速发展以及成本的快速下降,宏基因组高通量测序技术(metagenomic next generation sequencing,mNGS)的临床应用为解决上述问题带来了曙光[1, 2]。本文从mNGS技术原理与主要流程、临床转化与应用进展、数据库与分析系统构建要点和本地化实验室平台建设四个方面作概述。
一、mNGS技术原理与主要流程
mNGS技术无需培养,直接抽提感染标本中的核酸(DNA或RNA)进行高通量测序,通过微生物物种的序列数据库进行比对分析,确定标本中微生物的种类和丰度。与传统临床的实验室检测方法相比,mNGS可以全面覆盖上万种病原体,无偏向性快速鉴定细菌、真菌、病毒、结核、非结核分枝、支原体、衣原体和寄生虫等多种病原微生物。
mNGS检测流程主要分为5个步骤:样本前处理、核酸提取、文库制备、上机测序、生信分析与报告解读,全流程耗时14-20小时(图1、3)。目前,在样本前处理和核酸提取环节,需要面对临床标本中人源细胞占比不同、微生物核酸提取效率不同等难点,优化及稳定mNGS检测性能;在生信分析和解读环节,需要面对数据库建设全面性与精准性、技术解读和临床解读复合型人才培养等挑战。在临床转化与应用过程中,由于mNGS技术流程复杂,需要建立标准的实验室和制定标准化检测方法,并建议相关实验室在完成性能确认的情况下进行临床应用。
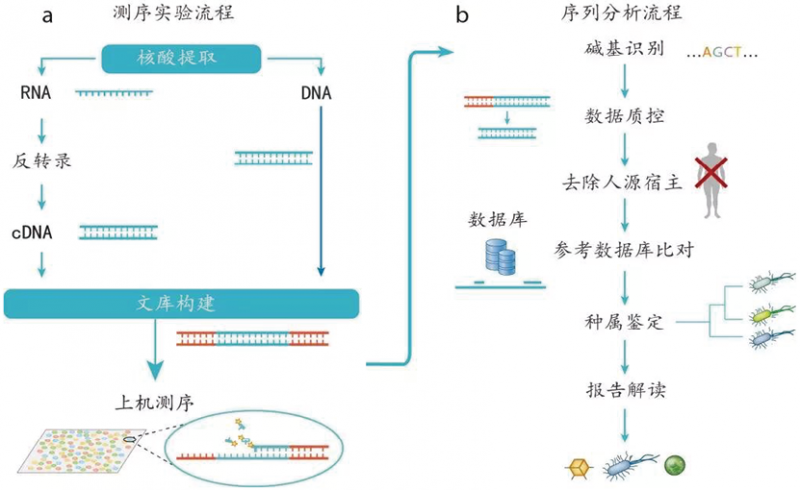
图1. mNGS检测全流程示意图
二、mNGS临床转化与应用进展
2014年,Wilson等首次应用mNGS诊断一例联合免疫缺陷患者钩端螺旋体病, 由此开启mNGS临床应用时代[3]。7年来,mNGS临床应用已从病原体鉴定(新发、罕见、特殊病原体报道),临床大样本数据研究(方法学性能、病原谱、患者预后),迈向区分定植与感染、鉴定耐药和毒力、分析宿主转录组学的感染精准诊疗之路。
1. mNGS应用于新发/罕见病原体鉴定:mNGS对于检测新发、罕见、特殊病原体具有显著的优势。2019年底,mNGS首先从不明原因肺炎病例中发现一种未被报道过的病毒,通过深入分析,耗时仅4天鉴定出新型冠状病毒[4]。另外,mNGS不仅对罕见、特殊病原体的临床诊断具有重大价值,如mNGS辅助诊断猪疱疹病毒的跨界传播、罕见肝结核、鹦鹉热肺炎等诸多疑难感染病例[5-7],而且对具有强传染性的罕见病原体具有重要公共卫生疾病防控意义,如2019年两名来自内蒙古的在京肺炎患者,mNGS提示鼠疫耶尔森菌感染,快速为疾病诊疗和防控提供了方向[8]。
2. mNGS应用于临床疑难重症感染诊治:mNGS与常规检测方法联合可以提高病原学诊断概率,有助于改善患者预后,减少社会经济损失。Blauwkamp等纳入350例脓毒症患者,发现mNGS联合血培养和其它检测方法可以显著提高病原体检出率到52%[9]。钟南山团队纳入142位肺炎患者,发现mNGS联合传统微生物检测方法,可显著提高病原体检出率到90.7%,检测结果可用于指导针对性用药或调整经验性治疗方案[10]。谢菲等关注mNGS对治疗的收益评估,入组159名社区获得性肺炎患者,发现相比于常规微生物检测组,mNGS辅助早期病原学诊断,患者症状缓解率从7%提升到55.9%[11]。谢云等探讨mNGS早期提供病原学证据和ICU患者生存情况之间的关系,对178例重症肺炎患者进行回顾性研究,发现mNGS为临床的早期明确诊疗提供线索,临床据此调整患者治疗方案后,患者28天和90天生存率均出现提高[12]。
3. mNGS在病原耐药与毒力分析的初步应用:mNGS可以实现微生物耐药/毒力基因与人体转录组分析。Langelier等入组92例下呼吸道重症肺炎患者,创新性的整合微生物组和宿主转录组结果,构建感染与非感染的mNGS结果判定模型[13]。Chiu&Miller提出mNGS可用于检测临床病原体耐药基因和毒力因子[14],Hasman等通过mNGS检测尿液样本中病原体及其耐药基因,发现病原体耐药性不仅符合基于纯培养物的全基因组测序结果,也与药敏试验结果一致[15]。张文宏团队纳入187例肺炎患者,用宿主转录组信息筛选出36个差异表达基因,从宿主免疫应答角度预测肺炎患者是否为新冠病毒感染相关[16]。
4. mNGS临床与检验应用专家共识:在急诊领域,2019年国内专家发布了《宏基因组分析和诊断技术在急危重症感染应用专家共识》[18]。作为首篇mNGS临床应用专家共识,其对mNGS临床应用场景、研究进展、基本流程、挑战和未来发展方向做出详细概述。
在重症领域,2020年5月发布了《宏基因组测序技术在中重症感染中的临床应用专家共识》[19]。该共识围绕检测、规则和应用等方面阐述了基于mNGS技术在临床中重症感染患者病原学诊断中的应用,并对其未来在临床应用改进方面提出了展望。
在感染领域,2020年11月张文宏教授团队发布了《中国宏基因组学第二代测序技术检测感染病原体的临床应用专家共识》[20]。该共识就mNGS的临床应用范围、样本采集、分析解读和诊断效能等进行了证据总结,首次针对不同感染症候群、不同病原体的适用范围给出了相应的推荐意见,建立了中国感染病原体宏基因组学检测的标准规范。
在检验领域,在2021年,中华检验医学杂志陆续发布了王成彬教授,王辉教授等专家牵头撰写的《高通量宏基因组测序技术检测病原微生物的临床应用规范化专家共识》,《宏基因组高通量测序技术应用于感染性疾病病原检测中国专家共识》,《宏基因组测序病原微生物检测生物信息学分析规范化管理专家共识》等三篇专家共识[21-23],对mNGS技术全流程质控和实验室管理提出要求,并对该技术的临床应用提出指导意见。
三、mNGS数据库与生信分析系统构建
1. 生信分析流程:数据库构建进展与挑战。数据库是mNGS干实验分析流程中的核心组成部分,直接影响着mNGS检测的病原微生物种类数、准确度及分析性能等,具体因功能可以为人源参考基因组数据库、微生物基因组数据库[24, 25]。微生物数据库基于功能或临床报告需求可以进一步细分为耐药/毒力因子数据库,背景菌数据库、微生态菌群数据库等。

图2. mNGS三级微生物数据库
人源数据库的构建除纳入最新的国际通用人类参基因组数据库GRCh38.p13外,也需要考虑到地域、性别特征等,如国内建议补充中国人基因组如YH2.0基因组甚至最新的北方汉族参考基因组(NH1.0),包含不同性别的其它人类参考基因组等[26-28]。另外,人体转录组数据库RefSeq、线粒体基因组数据库MITOMAP等数据库可以有效去除测序序列中的人体转录组与线粒体核酸序列[29, 30]。通过与包含上述关键要点、构建相较全面的人源数据库进行比对,可以有效去除测序序列中庞杂的人源核酸信息,筛选出包含疑似致病微生物的核酸序列。
微生物数据库基因组主要来源于公共数据库如NCBI、Genbank等。大型数据库微生物基因组数据多样但通常污染严重,并未同时兼具全面性与高效的分析性能,而小型或分类别数据库如ViralZone、FDA-ARGOS通常物种单一或纳入的物种类别不全面[31, 32]。因此,基于公共数据库筛选高质量,组装完整的物种或菌株基因组,构建兼具全面与精准的数据库存在挑战。
除审慎筛选各物种高质量、组装完整的代表基因组外,还需考虑区域、时间等维度相同物种不同菌株的基因组差异,针对临床重要病原体构建更加精准的临床应用级别数据库。因此,已有相关专利提出通过构建物种融合基因组的方法[33]。该方法整合了物种所有可靠菌株的差异序列,保留丰富物种菌株信息,能有效避免病原宏基因(转录)组检测结果的假阳性、假阴性,提高检测准确度;同时也去除了物种内的冗余序列,大大减少数据库的数据量,减少分析计算资源的需求,缩短分析时间,降低分析成本。
耐药/毒力因子数据库主要基于已有的公共数据库如CARD、VFDB等[34, 35],通过与微生物耐药/毒力数据库比对,可以分析核酸序列中存在的耐药/毒力基因,进一步满足临床诊治需求。考虑数据库中绝大部分耐药基因临床意义不明,自然环境中也可存在耐药基因,因此耐药基因报告需要从序列可靠性、临床意义等多个维度考量,区分基因型与表型。
2. 报告解读流程:原则与要点。相较于其它传统检测方法,mNGS作为颠覆式创新技术所面临的挑战更为巨大[36]。破壁、核酸提取、文库构建、测序与生信分析等任一环节的微小变动将会影响mNGS的检测结果的准确性。例如核酸提取过程中,不可避免引入了来源于提取试剂盒的多种试剂工程菌,部分试剂盒中甚至含有微量的临床常见病原体如嗜麦芽窄食单胞菌、铜绿假单胞菌等,而且试剂工程菌的种类与个数因提取试剂盒而有所差异[37]。
因此,关于mNGS的报告,最新的专家共识建议准确识别试剂工程菌,通过构建背景菌数据库、基于阴性对照大数据建立基线等方式,尽可能排除实验过程中试剂引入的微生物,将样本中含有的病原体核酸信息真实呈现给临床[20]。另外,对于来自于人体皮肤/呼吸道的正常微生物菌群,需要加以区分,在免疫缺陷/低下患者重点关注其致病的可能。
mNGS数据下机图形化展示后,需要进行两个解读,即技术解读与临床解读。技术解读由mNGS技术从业人员、微生物学专家或行业交叉人才完成,过程重点通过病原体特点、序列数、相对丰度、鉴定置信度、阴性/阳性对照以及部分临床信息或通过与临床专家沟通等等判断样本中是否存在疑似病原体核酸序列,从结果层面确认单个/多个菌存在的可能性。临床解读主要有来自于感染相关各科室的专家完成,必要情况下需要技术解读人才的协同完成,过程重点为结合患者病史,临床症状,影像学与其它微生物学证据等判断患者是否感染某种/多种病原体,是否需要考虑报告的耐药基因/毒力因子等,做出诊治判断后并持续跟踪。虽然现阶段行业在报告解读层面尚未有统一的共识,但准确报告、准确解读并形成密切的沟通闭环是mNGS技术从业人员、微生物学专家与临床专家共同努力的方向。同时通过类似中国mNGS解读联盟的组织加速mNGS跨学科人才培养,规范mNGS临床应用是行业共同的目标。
四、mNGS本地化实验室平台建设
经过5年的技术发展与临床转化研究,mNGS检测正在回归临床检验,进一步走向规范化与自动化。2021年10月28日,国家卫生健康委医院管理研究所发布《关于印发“提高住院患者抗菌药物治疗前病原学送检率”专项行动指导意见的函》,病原基因组测序被列入常见病原学检验项目目录,并指明了宏基因组学测序的方法学优势在于高通量序列分析,通过序列比对溯源基因库实现病原体鉴定,以及发现未知病原体。
1. mNGS自动化平台建设走向全程化、模块化、智能化:生化免疫检验已经发展到了“样本进,结果出”的全自动化流水线阶段,而临床微生物检验的自动化仍处于更早的阶段,其关键因素在与微生物样本类型众多,处理流程繁琐,报告结果复杂。作为感染性疾病诊断的新方法学,mNGS自动化平台建设的理念和方向应为:“全程化,模块化,智能化”。
(1)全程化:面对包括血液、呼吸道、神经系统、组织以及其他局灶部位样本,进行DNA或RNA检测流程,mNGS检测应该建立起从样本质量到测序数据质控的覆盖全流程自动化检测与质控环节。
(2)模块化:mNGS处在方法学快速发展和深化过程中,其中的关键模块包括样本前处理,自动化核酸提取仪,自动化建库仪,自动化测序仪,与自动化生信分析与智能报告系统等五大模块,在提取与建库环节有两步定量过程,而在去宿主,随机/靶向建库环节都将进一步优化与突破。因此,当前的mNGS平台建设应该在各个模块选择最适宜的自动化设备,才能适应未来方法学的进一步发展。同时,模块化的自动化流程,更加有利于实验室处理20-100个样本的灵活通量,保证检测效率与时间。
(3)智能化: mNGS的关键技术与瓶颈在于基因序列分析和报告解读,而要求每个实验室都配备专业的生物信息人员是不现实的。因此,本地化mNGS数据库与报告分析系统成为关键,要能够满足临床诊断需要,定期更新,并能够智能化的构建本实验室背景微生物数据库,计算判读阈值,并结合mNGS专家报告系统,辅助临床解读。
在“全程化,模块化,智能化”mNGS实验室建成后,有望实现以2-3名技术人员,每天处理60-100个样本,实现最快13.5小时,平均20小时的检测报告时间,在本地进行规范而高效的mNGS检测。
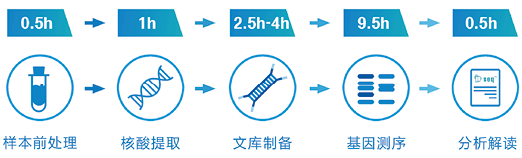
图3. mNGS主要步骤及时间
2.性能确认与质量评估:由于mNGS测序全流程步骤多,操作复杂,因此在临床和实验室开展应用前,需遵照CLIA和CAP实验室管理相关指导文件,进行mNGS检测技术的性能确认[39]。性能确认方案主要分为以下几个部分:检测样本类型和病原体范围,mNGS生物信息分析流程建立,生物信息分析流程性能确认参考盘制备,生物信息分析流程性能确认指标,mNGS实验流程建立,mNGS全流程性能确认参考盘制备,mNGS全流程性能确认,和mNGS全流程标准操作作业书(SOP)建立。
2021年国家卫健委临检中心启动了下呼吸道宏基因组学检测室间质评研究,本次活动从检测分析敏感性、特异性、报告解读及近缘物种的鉴定等多个方面考核了国内实验室mNGS检测的能力,共有超过90家医院与企业实验室参与,仅有55.6%的实验室结果合格, 其中5家单位满分[38]。因此可见,选择质量可靠,因地制宜的mNGS平台方案,是本地化实验室建设的关键。
五、总结与展望
mNGS经历了过去5年的蓬勃发展,已经在新发病原体鉴定与疑难重症诊治方面发挥了巨大的作用,并初步走向规范检验。未来5-10年,mNGS将进一步发展,与宏转录组学结合,通过随机测序与靶向测序技术并用,实现病原体鉴定分型与相对定量,耐药基因与毒力因子分析,宿主转录组学与免疫应答分析,从病原、药物和宿主三个维度进行病原检测与感染诊断分析,进一步的改变感染性疾病分子诊断格局,提升感染精准医学。
参考文献
Dekker JP. metagenomics for Clinical Infectious Disease Diagnostics Steps Closer to Reality[J]. J Clin Microbiol, 2018, 56(9), e00850-18.
Couto N, Schuele L, Raangs EC, et al. Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens[J]. Sci Rep-UK, 2018, 8(1), 13767.
Wilson MR, Naccache SN, Samayoa E, et al. Actionable diagnosis of neuroleptospirosis by next-generation sequencing[J]. N Engl J Med, 2014,370(25), 2408-2417.
Ren LL, Wang YM, Wu ZQ, et al. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study [J]. Chin Med J (Engl), 2020.
Ai JW, Weng SS, Cheng Q, et al. Human Endophthalmitis Caused by Pseudorabies Virus Infection, China, 2017[J]. Emerg Infect Dis, 2018, 24(6), 1087.
Ai JW, Li Y, Cheng Q, et al. Diagnosis of local hepatic tuberculosis through next-generation sequencing: Smarter, faster and better[J]. Clin Res Hepatol Gas, 2018, 42(3), 178-181.
Li N, Li S, Tan W, Wang H, et al. metagenomic next-generation sequencing in the family outbreak of psittacosis: the first reported family outbreak of psittacosis in China under COVID-19. Emerg Microbes Infect. 2021 Dec; 10(1): 1418-1428.
Wang YM, Zhou L, Fan MG, et al. Isolated Cases of Plague-Inner Mongolia-Beijing, 2019[J]. China CDC Weekly, 2019, 1(1): 13-16.
Blauwkamp TA, Thair S, Rosen MJ, et al. Analytical and clinical validation of a microbial cell-free DNA sequencing test for infectious disease[J]. Nature Microbiology. 4, 663-674 (2019).
Zhan Y, Xu T, He F, et al. Clinical evaluation of a metagenomics-based Assay for Pneumonia Management. Frontiers in Microbiology. 2021 ;12:751073. DOI: 10.3389/fmicb.2021.751073.
Xie F, Duan Z, Zeng W, et al. Clinical metagenomics assessments improve diagnosis and outcomes in community-acquired pneumonia[J]. BMC Infectious Diseases, 2021, 21(1).
Xie Y, Du J, Jin W, et al. Comparison the pathogen diagnosis of severe pneumonia by using next generation sequencing and traditional detection methods, China, 2010-2018[J], Journal of Infection, 2018.
Langelier C, Kalantar KL, Moazed F, et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc Natl Acad Sci U S A. 2018 Dec 26;115(52):E12353-E12362.
Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet 20, 341-355 (2019).
Hasman H, Saputra D, Sicheritz-Ponten T, et al. Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples[J]. J Clin Microbiol, 2014, 52(1), 139-146.
Zhang HC, Ai JW, Yang WJ, et al. metatranscriptomic Characterization of COVID-19 Identified A Host Transcriptional Classifier Associated With Immune Signaling[J]. Clinical Infectious Diseases 73(3), 376-385 (2020).
Infectious Disease Next Generation Sequencing based Diagnostic Devices: Microbial Identification and Detection of Antimicrobial Resistance and Virulence Markers. Draft Guidance for Industry and Food and Drug Administration Staff, 2016.
宏基因组分析和诊断技术在急危重症感染应用专家共识组. 宏基因组分析和诊断技术在急危重症感染应用的专家共识[J]. 中华急诊医学杂志, 2019, 28(2):5.
宏基因组学测序技术在中重症感染中的临床应用共识专家组, 中国研究型医院学会脓毒症与休克专业委员会, 中国微生物学会微生物毒素专业委员会,等. 宏基因组学测序技术在中重症感染中的临床应用专家共识(第一版)[J]. 中华危重病急救医学, 2020, 32(5):6.
《中华传染病杂志》编辑委员会. 中国宏基因组学第二代测序技术检测感染病原体的临床应用专家共识[J]. 中华传染病杂志, 2020, 38(11): 681-689.
中华医学会检验医学分会. 高通量宏基因组测序技术检测病原微生物的临床应用规范化专家共识[J]. 中华检验医学杂志, 2021, 43(12):15.
中华医学会检验医学分会临床微生物学组, 中华医学会微生物学与免疫学分会临床微生物学组, 中国医疗保健国际交流促进会临床微生物与感染分会. 宏基因组高通量测序技术应用于感染性疾病病原检测中国专家共识[J]. 中华检验医学杂志, 2021, 44(02): 107-120.
中华医学会检验医学分会. 宏基因组测序病原微生物检测生物信息学分析规范化管理专家共 识[J]. 中华检验医学杂志, 2021, 44(9): 799-807.
Ye SH, Siddle KJ, Park DJ, et al. Benchmarking metagenomics Tools for Taxonomic Classification[J]. Cell, 2019, 178(4): 779-794.
Breitwieser FP, Lu J, Salzberg SL. A review of methods and databases for metagenomic classification and assembly. Brief Bioinform. 2019 Jul 19; 20(4): 1125-1136.
Wang J, Wang W, Li R, et al. The diploid genome sequence of an Asian individual. Nature. 2008 Nov 6; 456(7218): 60-5.
Du Z, Ma L, Qu H, et al. Whole Genome Analyses of Chinese Population and De Novo Assembly of A Northern Han Genome. Genomics Proteomics Bioinformatics. 2019 Jun; 17(3) :229-247.
Pendleton M, Sebra R, Pang AW, et al. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat Methods. 2015 Aug; 12(8): 780-6.
O'Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016 Jan 4; 44(D1): D733-45.
Brandon MC, Lott MT, Nguyen KC, et al. MITOMAP: a human mitochondrial genome database--2004 update. Nucleic Acids Res. 2005 Jan 1; 33(Database issue): D611-3.
Masson P, Hulo C, De Castro E, et al. Nucleic Acids Res. 2013 Jan; 41(Database issue) D579-83.
Sichtig H, Minogue T, Yan Y, et al. FDA-ARGOS is a database with public quality-controlled reference genomes for diagnostic use and regulatory science[J]. Nat Commun, 2019, 10(1): 3313.
广州微远基因科技有限公司. 病原微生物基因组数据库及其建立方法: 中国,CN 110473594 A. 2019-11-19;
Alcock BP, Raphenya AR, Lau TTY, et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020 Jan 8;48(D1):D517-D525. doi: 10.1093/nar/gkz935. PMID: 31665441; PMCID: PMC7145624.
Liu B, Zheng D, Jin Q, et al. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019 Jan 8;47(D1):D687-D692. doi: 10.1093/nar/gky1080. PMID: 30395255; PMCID: PMC6324032.
Mitchell SL, Simner PJ. Clin Lab Med. 2019 Sep;39(3):405-418. doi: 10.1016/j.cll.2019.05.003. PMID: 31383265.
Salter SJ, Cox MJ, Turek EM, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014 Nov 12;12:87. doi: 10.1186/s12915-014-0087-z. PMID: 25387460; PMCID: PMC4228153.
2020年全国下呼吸道感染宏基因组测序室间质量评价预研活动结果报告
Burd, E. M. Validation of laboratory-developed molecular assays for infectious diseases. Clin Microbiol Rev23, 550-576, doi:10.1128/CMR.00074-09 (2010).


