


宏基因组测序病原微生物检测生物信息学分析规范化管理专家共识解读

杨斌,博士,微远基因临床生信科学家。北京大学博士,中国医学科学院病原生物学研究所梅里埃实验室博士后,美国科学院院士James. M. Tiedje实验室访问学者,曾任illumina北区应用科学家和大中华区微生物NGS应用专家。在呼吸道宏基因组组学数据分析、耐药及毒力基因生信分析上有近十年的工作经验。参与研究工作发表在Am J Respir Crit Care Med,mSystems等杂志上。
【摘要】宏基因组高通量测序技术(metagenome next-generation sequencing,mNGS)是一种无偏倚的病原体检测技术,已被广泛应用在各种临床感染相关样本的检测中。中华医学会检验医学分会在鲁辛辛主任和王成彬主任的带领下,撰写了《宏基因组测序病原微生物检测生物信息学分析规范化管理专家共识》。本文将就共识中涉及到的生物信息学概念和生物信息分析流程进行深入讲解和解读,以期协助临床和检验工作者更好地理解和应用专家共识。
【关键词】宏基因组高通量测序技术;生物信息学分析;专家共识;解读
宏基因组高通量测序技术(metagenome next-generation sequencing,mNGS),是一种基于宏基因组学和高通量测序技术,可检测并分析各种临床来源样本中所有已知及未知的病原体(包含细菌、真菌、病毒、寄生虫、支原体/衣原体等)。但由于mNGS全流程步骤多,操作复杂,导致目前众多mNGS检测体系选择的技术选型和分析方法各不相同,缺乏一定地可比性[1-4]。所以针对mNGS是否能在临床中开展和应用,存在较大的争议。其中在如黑匣子般的生物信息分析领域,规范化的问题尤为突出[5-7]。因此中华医学会检验医学分会在鲁辛辛主任和王成彬主任的带领下,撰写了《宏基因组测序病原微生物检测生物信息学分析规范化管理专家共识》。该共识一经发出,引起行业内巨大的反响和热议。同时由于专业度较高和受限于文章篇幅,导致一部分起步较晚的临床和检验老师难以完成理解共识中的内容。为了让文章能更多地造福临床和检验工作者,笔者及其工作团队尝试去针对《宏基因组测序病原微生物检测生物信息学分析规范化管理专家共识》(下简称共识)中的内容基于笔者及工作团队有限的学识进行展开和解读。如果文中存在不足或理解上的错误,也恳请各位专家和同道批评指正。
一、基本概念的解读
1. 什么是文库、序列和读长?二代测序(NGS)其中一个特征就是需要构建文库。需要测序的基因组核酸被打断成短片段(100-500bp)之后,被加上能够被高通量测序仪所识别的接头序列[2]。如图1所示,接头的最外侧是用于和测序芯片上的寡核苷酸进行偶联,中间的部分是用于区分样本的barcode序列,最里面则是测序所需要的测序引物。被打断的基因组短片段成为插入片段,即我们的检测目标片段[1]。一个文库分子对应就是我们测序数据中的一条序列(read)。二代测序还有一个特征则是会将一个文库分子从5’端测定一次,从3’端再测定一次,也是大家熟知的双端测序。所以双端测序的策略会产生两条序列,5’端开始的,称为Forward read, 3’端开始的,成为Reverse read。但由于测序时间的因素限制,mNGS检测大多采用单端的测序方式。读长指的是从开始(5’/3’端)到结束一共测定了多少个碱基。所以,当插入(目标)片段长于300bp的时候,即使双端均测定150bp,也无法测通[8]。需要注意的是,已有的mNGS临床研究发现,50-75bp的读长,足够用于mNGS的分析[9, 10]。
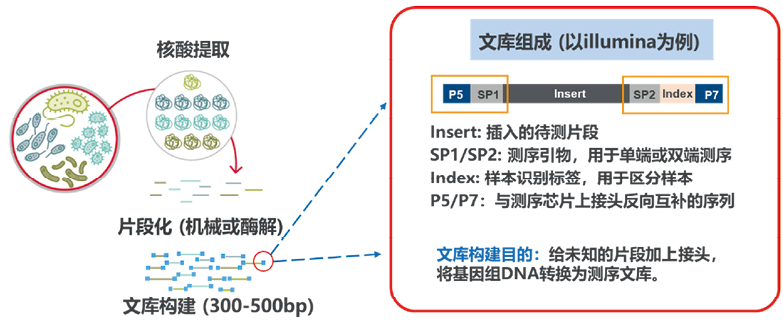
图1. 二代测序文库结构示意图
2. 绝对丰度和相对丰度:相对丰度、绝对丰度、每百万序列数和每千万序列数是临床医生在面对mNGS报告时,经常能够遇到的生物信息学概念。在这里,笔者希望给广大临床工作者从两个方面进行解读:(1)为什么会产生这些概念?(2)这些指标在什么情况下有临床价值,能够指导临床进行诊断或治疗?首先回答第一个问题,为什么会产生这些概念?如图2所示,高通量测序是一个随机抽样的检测过程,并不是样本中所有的核酸都会被尽数检测到。因此每个样本的高通量测序数据只是样本的一部分。例如一管样本中存在1ng的文库分子,那如果片段大小平均都在350bp左右,它将有2.4×1015个文库片段。根据1.1中的解释,一个文库片段对应的就是测序数据中的一条序列。如果都进行测序,将会产生2.4×109M reads。但实际上我们只会进行5-100M reads的测定。所以除非我们穷举这管样本中的所有文库分子(但基本是难以实现),否则我们检测到的人源序列还是微生物序列数,更多反应的是其在样本中的占比情况。因此,如果该微生物在样本中占比越大,它被测到的概率越高,它的序列数也就随之越高。另外一方面,如果样本总测序序列数越多,它能检测的低比例微生物,则越多。例如一个微生物的核酸在样本中占比是一千万分之一(即1/10M),则需要针对这个样本检测超过10M reads时,它才有可能被检测到[11]。所以,生信专家共识中提到的绝对丰度,就是指该微生物文库片段/序列在样本中的占比。而每百万/千万序列数则是将绝对丰度乘以一百万或一千万,意为(均一化到)一百万/千万总测序序列中,能检测到多少该微生物的序列数。还有一些研究团队,为了更加贴合宏基因组临床应用专家共识,将这个均一化的基数,设置为两千万(20M reads)[9, 12]。相对丰度则是指该微生物序列在所有微生物序列中的占比。在肠道微生物组的研究中,一般认为在种群中丰度占据绝对地位的微生物会发挥重要的作用。因此,考虑相对丰度的分析方式也被随之代入到了mNGS领域[13, 14]。但由于不同的微生物基因组大小不一、破壁难度不一(真菌>细菌>病毒)[1, 2, 6]。因此,也有专家把不同类型的微生物单独计算它们的相对丰度(如细菌A只考虑在细菌中的相对丰度)[4]。所以在讨论相对丰度的时候,各位临床老师需要注意分母指的是什么,这可能会影响计算的结果。
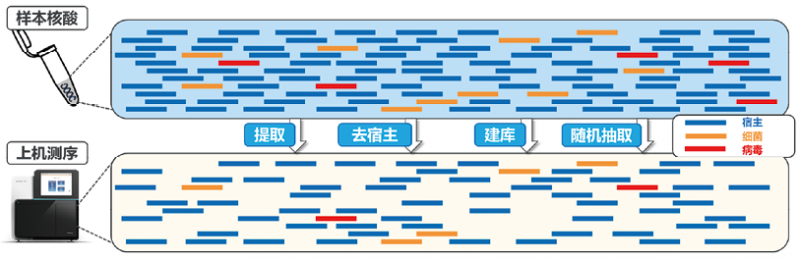
图2. 二代测序随机抽样示意图
第二个问题,这些指标在什么情况下具有临床价值,即提示临床该微生物可能是致病原。这也是经常会引起临床老师的困惑。这是mNGS临床应用中较为前沿的科学问题。笔者在第一个问题中花了较大的篇幅,则是为了说明为什么回答这个问题存在一定的难度。因为微生物的占比会随着人源细胞的变化和其他微生物的载量变化而变化。即使相同载量的微生物,也可能在不同的样本中呈现不同的绝对丰度、相对丰度和每百万序列数。目前已经有一些研究团队尝试去解决该难题,如加州大学旧金山分校医学院和广州呼研院团队使用RPM和RPM在大样本中排名作为构建阈值的指标,筛选出阳性判断阈值,用于指导免疫缺陷儿童和成人的肺部感染病原体诊断。笔者相信,随着更多的mNGS临床研究和技术方法的升级,具有临床指导意义的mNGS检测阈值一定会出现[15, 16]。
3. 基因/基因组覆盖度和平均测序深度:基因/基因组覆盖度和平均测序深度主要是为了防止物种在注释的过程中存在错误比对的生物信息学评价指标。如图3所示,对于图3A中的新冠基因组,比对上的序列基本覆盖了整个新冠的基因组,但每个碱基只被覆盖了3-4次(覆盖度为:平均测序深度为)。相反,图3B中的序列只在几个简单的位置出现,但每个位置都有超过100X的测序深度。尽管A图和B图的样本都有相近的序列数比对上新冠,但A图所代表的样本比B图更有可能存在新冠病毒,而B图则更有可能是因为PCR产物或新冠核酸检测中常用的阳性质控品引入的污染。因此,理论上来说,比对上的序列在该物种的基因组上分布越均匀,支持该物种基因组存在在样本中的证据强度越高。除此之外,美国病理学会在2017年发表的关于mNGS性能确认的共识中提到,出现这个情况的另外一种原因,是数据库中缺少该物种的代表基因组,这是错误的比对结果[3]。这个现象会在新发病原体出现的时候出现,如新冠。物种序列仅依靠同源性区域被比对上,其他绝大部分序列都无法比对上。因此,重要的生物信息学指标对于判断结果是否可靠是不可或缺的[5, 6, 17-19]。
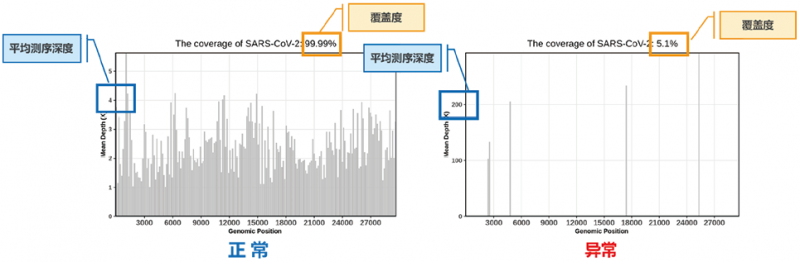
图3. 新冠病毒基因组覆盖度示意图
4. 碱基质量值:碱基质量值储存在FASTQ文件中每条序列的第四行。许多临床或检验的老师会认为这是一些乱码。实际上这是计算机的语言,通过ASCII(American Standard Code for Information Interchange)码。这是将两位数字转化为一位字母的编码方式。由于计算机特殊的工作原理,每增加一个数位(例如从个位数变成十位数加个位数),存储将增加一倍。计算机学家发明了这样一套编码的计算方法,缩减了一倍的储存空间。同时,原有的数字展示的是该碱基的测序错误率,例如Q30就是千分之一的错误率,Q20就是百分之一的错误率[20]。但细心的读者可能会发现,明明我只是测了一次这个碱基,它的测序错误率是从哪里来的呢?实际上,高通量测序仪并不是真正去测定这个碱基的错误率,而是通过一系列的光学参数,推断这个碱基的错误概率。高通量测序仪厂商测定大量已知序列的参考基因组,构建起光学参数和碱基错误率的数学模型,因此可以通过之后每次测定的光学参数代入到已有的数学模型中,求得该次的测序错误率。所以,不同厂商的测序错误率或者是Q值是不能也不应该相互比较的,因为每家构建的数学模型都不尽相同[21]。真正评价不同高通量测序仪的性能,应该仿照ABRF(Association of Biomolecular Resource Facilities)使用明确序列的标准品进行测定,比较突变率/错误率,才能真正横向比较不同测序仪的测序准确性。从ABRF发表在Nature Biotechnology上的研究看到,目前国产的测序仪准确性已经不亚于国际一流品牌的测序仪[22]。
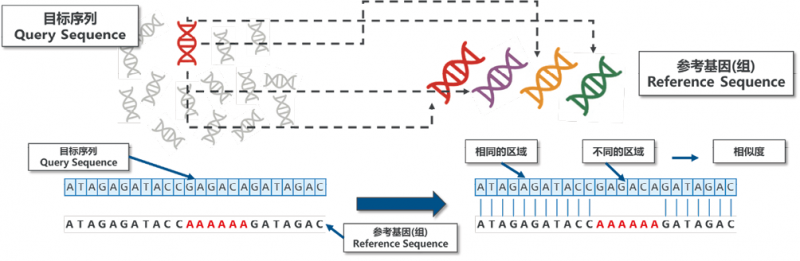
5. 什么是比对?比对有什么作用?序列比对(Sequence Alignment)是生物信息学的基本工具,主要作用是用于找到两条核酸序列或者是氨基酸序列相似的地方和不同的地方。通过计算相似的占比,可以得到两条序列的相似度,是现今生物信息学发展的基石(图4)。mNGS分析就是通过与已经构建好的数据库进行比对,找到最为相近的参考基因组序列,根据参考基因组序列的物种分类地位,注释该序列的物种分类。与之相对应的是非比对(Alignment-free),这是两种找到近源序列的分析方法。各自有自身的优势和劣势。比对的优势在于准确性高,但是需要高配置的硬件和较长的计算时间。非比对的优势在于快速,但劣势则是准确性略差[5]。
二、基本技术方案的解读
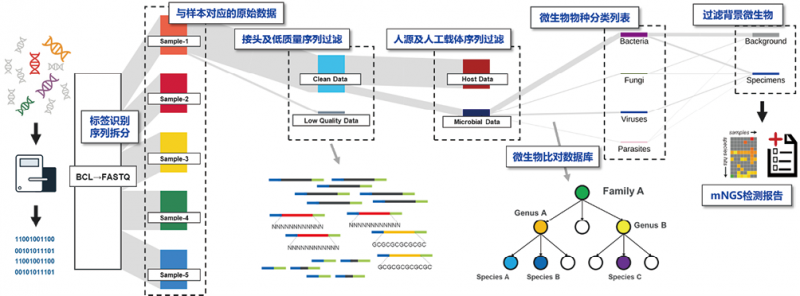
《共识》中明确提出,mNGS的分析流程涵盖了标签识别、序列拆分、接头及低质量序列过滤、人源及人工载体序列过滤、微生物比对数据库、根据物种同源性百分比、按序列高低排序、微生物物种分类列表、结合样本类型和临床特征、过滤背景微生物和生成mNGS报告。笔者将一份样本从文库分子到mNGS报告制作了一张流程图,尝试阐释mNGS的关键步骤和数据流向(图5)。此外,由于这部分涉及比较多的生物信息学算法和概念,笔者仅选取部分在《共识》中并未充分展开的问题进行讨论和解读。
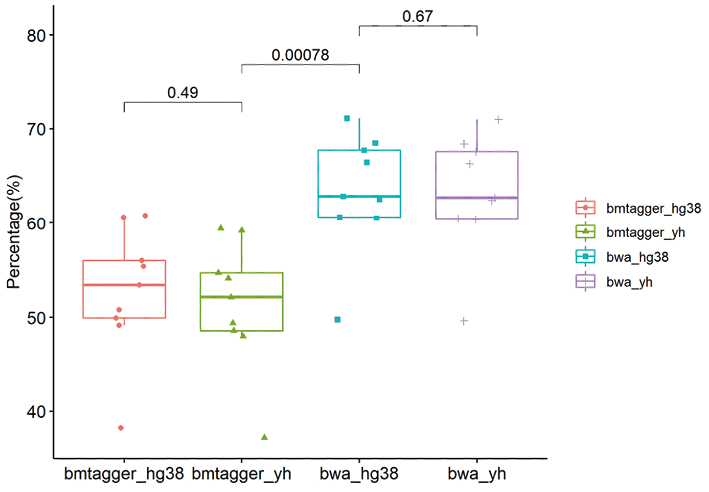
1. 人源序列过滤:《共识》中提及到,人源序列的残留会造成后续微生物序列分析的错误,因此有效地过滤掉人源序列既可以减少后续分析的错误,也可以加速微生物物种注释的时间。所以是数据前处理中极为重要的过程。笔者注意到,有一些mNGS的厂家过分夸大炎黄一号与hg19/38等高加索人的基因组差异在人源序列过滤中的作用。笔者非常同意加入炎黄一号作为人源序列去除的比对基因组。但过分强调基因组而忽略选择合适的算法,会是舍本逐末。如图所示,笔者曾做了一个小样本的测试,选择更为精细化的BMTagger[23]和BWA[24]进行人源序列过滤的对比分析。从结果上看(图6),在比对分析上更为精细的BMTagger所带来的差异,要远远高于所谓的人种基因组差异带来的影响。目前mNGS厂家鱼龙混杂,有些从业者利用临床和检验老师对生物信息学的不熟悉,刻意营销概念,也是需要警惕的。
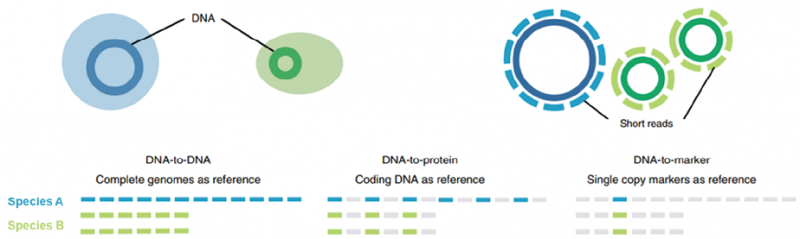
2. 物种注释和数据库构建:宏基因组是生物信息学分析中的皇冠,而物种注释则是这个皇冠上的珍珠。其准确性左右了整个mNGS检测的可靠性。目前物种注释的算法分为上文提及的比对方式和非比对方式。数据库则根据参考序列的特征,分为了全基因组、种特异的核酸序列和蛋白序列三种数据库类型(图7)[5]。算法和数据库不同类型的组合,形成了目前学界五花八门的宏基因组物种注释软件。《共识》中列举了几种最具有代表性的物种注释软件。根据已有的研究,基于种特异核酸序列作为参考数据库的软件,展现出了较高的特异性,但其敏感性则要弱于基于全基因组的分析软件。因此在特异性和敏感性的平衡上,需要对数据库进行一番精心的清洗和整理。而且《共识》指出,如何推动感染性疾病规范化诊疗、推动以疾病诊断为基础的检测试剂注册、推动以疾病分类建设微生物检验技术平台是今后应解决的首要问题。所以构建好的分析软件和数据库应当进行检测注册证申请,经过权威机构部门认可和验证的的分析软件和数据库,才是真正值得信赖和依靠的分析系统[16]。
3. 物种注释结果准确性判断和背景去除:物种注释结果准确性判断主要依靠一些重要的生物信息学参数,如上文提及的覆盖度、平均测序深度,还有其他指标如相对丰度和离散度等。如果在检测报告中展示过多这些生物信息学的概念,会给临床造成一定的困扰。检验工作者和生物信息工作者可以尝试共同开发出一套易于理解、简洁且科学的指标用于展示物种注释结果的准确性和可靠性。另外一方面,目前大量的研究表明,我们所使用的分子生物学试剂盒存在大量的背景核酸。因此,需要建立背景菌/试剂工程菌的列表,针对存在于试剂中的微生物设立阳性检出阈值(Cutoff value),防止假阳性的检测结果[25]。笔者也听到一些mNGS厂家提出,任何一份标本都能检测出病原体/微生物。这个概念是极其误导临床工作者的。任何一种方法学都存在最低检出限,这是检验所该有的理念和原则。同时,任何一个阈值都会造成一定程度的假阳性和假阴性,取决于阈值建立的目的是为了保证敏感性还是特异性[26]。目前国际上已有一些研究针对自身团队建立的实验方法设立阈值,检验工作者在建立实验室技术体系或者对引入的方法进行性能确认的时候,可以参考和学习[10, 17, 27]。
三、总结
《共识》还就生物信息分析流程性能确认和平台建设提出了非常详尽的规范和建议,已经涵盖了该方面所需阐述的内容。因此,本文将不再累述。有兴趣的读者可自行查阅和参考《共识》进行相关的技术体系构建和性能确认。本文针对《共识》中无法展开的几个生物信息学概念进行了解读和讨论,以及分析流程中涉及的关键问题进行了展开,旨在作为《共识》的补充,协助各位临床和检验工作者用好和理解《共识》,让《共识》发挥更大的作用,为感染领域、病原学领域和国家卫生健康领域的发展增添科技的新动能。![]()
参考文献
[1]Gu, W., S. Miller, and C.Y. Chiu, Clinical metagenomic Next-Generation Sequencing for Pathogen Detection. Annu Rev Pathol, 2019. 14: p. 319-338.
Chiu, C.Y. and S.A. Miller, Clinical metagenomics. Nat Rev Genet, 2019. 20(6): p. 341-355.
Schlaberg, R., et al., Validation of metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection. Arch Pathol Lab Med, 2017. 141(6): p. 776-786.
中华医学会检验医学分会临床微生物学组,中华医学会微生物学与免疫学分会临床微生物学组,中国医疗保健国际交流促进会临床微生物与感染分会, 宏基因组高通量测序技术应用于感染性疾病病原检测中国专家共识. 中华检验医学杂志, 2021. 44(No.2.).
Ye, S.H., et al., Benchmarking metagenomics Tools for Taxonomic Classification. Cell, 2019. 178(4): p. 779-794.
Greninger, A.L., The challenge of diagnostic metagenomics. Expert Rev Mol Diagn, 2018. 18(7): p. 605-615.
Bharucha, T., et al., STROBE-metagenomics: a STROBE extension statement to guide the reporting of metagenomics studies. Lancet Infect Dis, 2020. 20(10): p. e251-e260.
Head, S.R., et al., Library construction for next-generation sequencing: overviews and challenges. Biotechniques, 2014. 56(2): p. 61-4, 66, 68, passim.
Xie, F., et al., Clinical metagenomics assessments improve diagnosis and outcomes in community-acquired pneumonia. BMC Infect Dis, 2021. 21(1): p. 352.
Gu, W., et al., Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med, 2021. 27(1): p. 115-124.
Ebinger, A., S. Fischer, and D. Hoper, A theoretical and generalized approach for the assessment of the sample-specific limit of detection for clinical metagenomics. Comput Struct Biotechnol J, 2021. 19: p. 732-742.
Xie, Y., et al., Next generation sequencing for diagnosis of severe pneumonia: China, 2010-2018. J Infect, 2019. 78(2): p. 158-169.
Thaiss, C.A., et al., The microbiome and innate immunity. Nature, 2016. 535(7610): p. 65-74.
Lim, M.Y., et al., Comparison of DNA extraction methods for human gut microbial community profiling. Syst Appl Microbiol, 2018. 41(2): p. 151-157.
Zinter, M.S., et al., Pulmonary metagenomic Sequencing Suggests Missed Infections in Immunocompromised Children. Clin Infect Dis, 2019. 68(11): p. 1847-1855.
Zhan, Y., et al., Clinical evaluation of a metagenomics-based Assay for Pneumonia Management. Front Microbiol, 2021. 12: p. 751073.
Miller, S., et al., Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res, 2019. 29(5): p. 831-842.
Mitchell, S.L. and P.J. Simner, Next-Generation Sequencing in Clinical Microbiology: Are We There Yet? Clin Lab Med, 2019. 39(3): p. 405-418.
Lopez-Labrador, F.X., et al., Recommendations for the introduction of metagenomic high-throughput sequencing in clinical virology, part I: Wet lab procedure. J Clin Virol, 2021. 134: p. 104691.
Zhang, S., et al., Estimating Phred scores of Illumina base calls by logistic regression and sparse modeling. BMC Bioinformatics, 2017. 18(1): p. 335.
Chen, Z., et al., Highly accurate fluorogenic DNA sequencing with information theory-based error correction. Nat Biotechnol, 2017. 35(12): p. 1170-1178.
Foox, J., et al., Performance assessment of DNA sequencing platforms in the ABRF Next-Generation Sequencing Study. Nat Biotechnol, 2021. 39(9): p. 1129-1140.
Shen, Z., et al., Genomic Diversity of Severe Acute Respiratory Syndrome-Coronavirus 2 in Patients With Coronavirus Disease 2019. Clin Infect Dis, 2020. 71(15): p. 713-720.
Li, H. and R. Durbin, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009. 25(14): p. 1754-60.
Olomu, I.N., et al., Elimination of "kitome" and "splashome" contamination results in lack of detection of a unique placental microbiome. BMC Microbiol, 2020. 20(1): p. 157.
Colquhoun, D., An investigation of the false discovery rate and the misinterpretation of p-values. R Soc Open Sci, 2014. 1(3): p. 140216.
Greninger, A.L. and S.N. Naccache, metagenomics to Assist in the Diagnosis of Bloodstream Infection. J Appl Lab Med, 2019. 3(4): p. 643-653.


