


基因检测在痛风发病及诊疗中的应用价值

刘栩,博士、主任医师。现任职于北京大学人民医院风湿免疫科。目前担任北京大学风湿免疫学系委员,海峡两岸医药卫生交流协会风湿免疫专委会感染学组委员,北京市中西医结合学会风湿病专委会委员。负责“强直性脊柱炎专病门诊”,担任《International Journal of Rheumatic Diseases》《Clinical Rheumatology》及《PLoS ONE》等杂志审稿人。作为项目负责人,负责2项国家自然科学基金(30901319,31470039)、北京市科技计划(Z191100006619112)及首都特色临床项目(Z141107002514064)。2018年获得新疆医科大学第一附属医院“优秀援疆干部”,2011年获得《中华风湿病学杂志》优秀论文奖,2009年获得北京风湿病年会优秀论文奖。作为第一作者或通讯作者,发表5篇SCI论文,发表国内核心期刊杂志文章10余篇,参与6部专著编写,《风湿病诊断手册》副主编。

冯卫民,北京大学第二临床医学院2018级医学检验技术专业本科生。曾获得“北京大学社会工作奖”、“基础医学院优秀团员”、“医学科普大赛”三等奖、北京大学人民医院“大学生创新实验项目”二等奖等奖项。
痛风是一种由于单钠尿酸盐结晶在关节或非关节位置的沉积,诱发局部炎症反应和组织破坏的疾病[1]。痛风在全球范围内总体发病率较高,但在不同地区、不同性别和年龄人群间发病率的差异较大。痛风的发病受到多种环境因素和遗传因素的影响,其中血清尿酸盐浓度升高(高尿酸血症)是痛风发展的最重要危险因素[2]。在既往的研究中已经证实,如SLC2A9基因[3]、SLC22A12基因[4]、LRP2基因[5]等多个基因都与血清尿酸盐升高和痛风的发病有着重要的关系。因此对于痛风发病相关的基因检测在痛风的诊断过程中发挥着重要作用。此外,目前临床上对于痛风的治疗主要分为药物抗炎治疗和药物降尿酸治疗,前者常常包括口服糖皮质激素、非甾体类抗炎药等;而后者常常使用别嘌呤醇、非布司他等药物,但这些药物的使用过程中易引发的各种不良反应,给临床治疗带来了困难。其中尤其是别嘌呤醇引发的严重的皮肤不良反应(SCAR)最为严重,大量病例和研究表明,HLA-B*5801等位基因与这一不良反应呈现很强的相关性,且中国人群携带此基因的频率较高,可达到10%-20%[6]。因此基因检测对于痛风的治疗、指导临床用药也有着重要的临床意义。
一、痛风的临床表现
痛风的临床表现具有多样性、复杂性,其发展过程主要可以分为四个病生理阶段:高尿酸血症的发展、尿酸盐结晶的沉积、对于结晶沉积引发的急性炎症和以痛风石为特征的晚期疾病[7]。大多数痛风患者的临床症状为痛风性关节炎的反复发作,且多骤然发作并伴剧烈疼痛,具体表现为关节疼痛、关节肿胀、活动受限、关节压痛、肢体麻木、肢体发凉、肢体困沉、畏寒怕冷、关节潮红/发热、指端苍白等[8]。在没有治疗的情况下,痛风发作通常会在7-14天的时间内自限性消退,但痛风的复发难以预测,复发的可能性与高尿酸血症的严重程度有关[9],发作的诱因包括高嘌呤饮食、饮酒、关节创伤和急性疾病等。

图1. 患者手指指垫和手伸肌肌腱及脚趾滑膜囊内的尿酸盐沉积[10]
二、痛风的诊断标准
在显微镜下确认滑液或痛风石中的单钠尿酸盐晶体被认为是诊断痛风的金标准[11]。但在存在典型的症状和临床指标的情况下,也能高度确认诊断痛风。根据2015年ACR/EULAR的分类标准,高尿酸血症的诊断标准为:在正常嘌呤饮食状态下,非同日2次空腹血尿酸(SUA)浓度>420μmol/L,无论男女。而当下表中累计分值≥8分即为痛风。该诊断标准除充分考虑患者的临床症状及血清尿酸水平外,还纳入了MSU沉积及高频超声、双能CT等影像学检查结果,使痛风诊断的敏感性和特异性均得到显著提升[12]。
表1. 2015年ACR/EULAR的痛风分类标准[13]

三、痛风的流行病学
高尿酸血症和痛风在全球范围内的发病率很高,而且一项基于美国人群的研究显示,在过去20年间仍在不断增加[14]。一般来说,痛风的患病率为1%~4%。高龄和男性是全球关注的两个常见风险因素。在西方国家,男性痛风患病率(3%至6%)是女性(1%至2%)的2至6倍[15];2015年韩国痛风患病率男性为1.36%,女性为0.16%[16]。此外,男性患痛风的时间也比女性早,丹麦一项研究发现男性的平均发病年龄为65.3岁,女性为71.4岁[17]。痛风的患病率也随着年龄的增长而增加,此前的研究表明,在加拿大,2012年70-79岁男性和女性的痛风患病率为11.8%,而50-59岁人群为5.1%,<30岁人群<1%[18];在澳大利亚,2013年男性痛风患病率从25-29岁人群的0.2%到85岁以上人群的11.05%[19]。同时,在不同的地区和人群中,高尿酸血症和痛风的发病率差异较大。在世界范围内,大洋洲的痛风患病率最高,为13.9%;在欧洲2003-2014年期间痛风的患病率在1%-4%之间;而在中国成年人口中痛风的患病率为1.1%,但近年来这一比例在持续升高[20]。

图2. 全球痛风患病率估计值[20]
四、痛风的发病风险因素
痛风是一种与遗传因素和环境因素均密切相关的疾病。如下表2中,痛风发病的风险因素多种多样,高尿酸血症并不是痛风的唯一危险因素,不合理的饮食结构,如过多的海鲜、红肉、酒精摄入等以及一些内在风险因素如肥胖、慢性的肾脏疾病、特殊药物的摄入等外界因素都会导致更高的痛风发病率[21-23]。
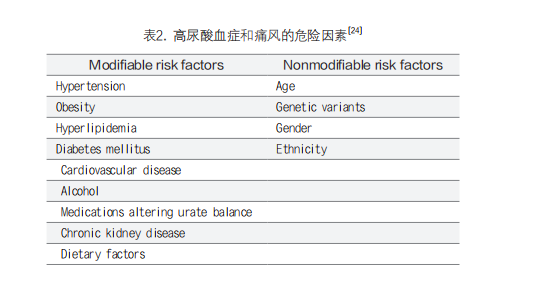
同时,多项研究表明,遗传因素对血清尿酸浓度影响很大,遗传力估计高达73%。全基因组(GWA)研究有9个基因座影响血清尿酸水平,分别是PDZK1、GCKR、SLC2A9、ABCG2、LRRC16A、SLC17A1、SLC16A9、SLC22A11、SLC22A12,且每个基因组内有一个或多个SNP与尿酸代谢密切相关,这些基因都将影响到痛风的患病率[25]。Anna Köttgen等人通过对140000名欧洲个体的临床数据分析,确定了28个与血清尿酸盐浓度相关的全基因组显著位点[26]。在一项日本学者主导的研究中,使用了71149名东亚人的数据进行分析发现,SLC2A9、ABCG2和SLC22A12基因与尿酸浓度存在明确的相关性[27]。在非洲裔美国人的研究中也有类似的结论,他们发现了3个对血清尿酸盐有显著性的基因座,分别是SLC2A12邻近的一个新基因座、SLC2A9和SLC22A12[28]。

如图3中显示,近年来的多次研究都证明了痛风和高尿酸血症的发病与多种易感基因密切相关,虽然对于不同人群的研究结果表明在不同人群中这些易感基因并不完全相同且多数基因的影响都十分有限,但是仍有个别基因,如SLC2A9、SLC22A12等在所有群体中都与痛风的发病密切相关。这些研究结果进一步说明了,在对于痛风的诊断过程中,基因检测将会发挥越来越重要的作用。
五、痛风的发病机制
在人体内尿酸主要来源是嘌呤物质的分解产物,而肝脏从头合成和补救途径中合成的嘌呤与消化道对嘌呤的吸收是体内嘌呤的主要来源。嘌呤代谢为尿酸后,肾脏大约排泄了其中的三分之二,而胃肠道排泄了剩余的三分之一。因此常见的高尿酸血症患者可能原因有肝脏代谢和细胞产生过量,肾脏和肠道排泄障碍以及肾脏重吸收增加,其中后两者在临床中更为多见。在肾脏尿酸排泄的过程中,近端小管会对尿酸进行重吸收,其中约90%重吸收入血,这一过程主要是通过细胞内阴离子交换转运蛋白完成的。其中,顶端膜上的三种尿酸转运蛋白URAT1/SLC22A12,GLUT9/SLC2A9和OAT4/SLC22A11介导尿酸盐从管腔转运到细胞内,而ABCG2、ABCG4和NPT1是分泌转运蛋白;在基底膜侧OAT1、OAT2和OAT3负责将尿酸盐转运到近端小管细胞内,而GLUT9(SLC2A9)调节尿酸盐的重吸收[30-38]。
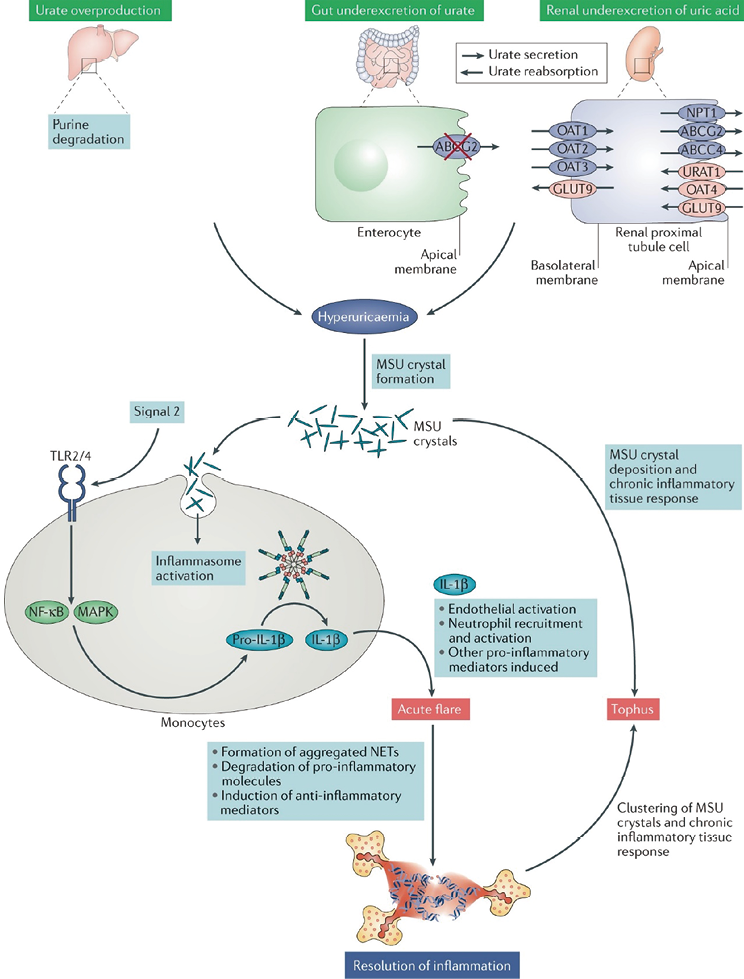
图4. 高尿酸血症进展为痛风[29]
当机体内尿酸盐代谢出现障碍形成单钠尿酸盐结晶后,会刺激巨噬细胞和单核细胞释放IL-1β,最终导致中性粒细胞和其它细胞聚集至单钠尿酸盐沉积部位导致炎症。在这一过程中,单核细胞、巨噬细胞和中性粒细胞发挥了重要的作用。
六、痛风的治疗
痛风发作时的优先事项是抑制疼痛和抑制关节炎症。建议早期给予抗炎治疗以快速抑制关节疼痛。这些药物中的一线选择是口服糖皮质激素、非甾体抗炎药和秋水仙碱。这三种药物的选择与患者的具体发病情况、是否有肾脏疾病和自身身体条件等因素相关[39, 40]。除了在痛风发作时给予对症治疗,患有高尿酸血症或痛风患者的降尿酸治疗更加关键。降尿酸治疗主要有三大类药物。第一大类药物的作用机制是抑制尿酸的生成,如别嘌呤醇、非布司他、托匹司他等等;第二大类药物的主要作用是促进尿酸的排泄,如苯溴马隆、丙磺舒等;第三类药物是通过催化尿酸盐转化为更容易溶于水且易于排泄的尿囊素,如拉布立酶、聚乙二醇化酶[41]。
虽然别嘌呤醇是治疗高尿酸血症和痛风的最常用的药物,但是别嘌呤醇也是导致严重皮肤不良反应的最常见原因之一,其中包括药物超敏反应综合征、史蒂文斯-约翰逊综合征(SJS)和中毒性表皮坏死松解征(TEN)。尤其是在HLA-B*58:01等位基因阳性的患者中,别嘌呤醇诱发不良反应的概率极高[6]。非布司他为选择性黄嘌呤氧化酶抑制剂,具有很强的特异性,应用该药会改善血管内皮细胞功能,抑制血小板聚集,减轻炎症反应,由于其具有高特异性,在临床治疗中不会影响其他嘌呤、嘧啶的代谢。同时非布司他能够通过肝肾双通道代谢,对于具有肾功能不全的患者更加有利,但是该药在临床使用中价格较贵且可能引发未明确的心血管事件[42]。因此在痛风治疗的临床用药上仍然需要更好的指导,基因检测在这一过程中有重要价值,当前与痛风治疗相关的基因研究主要集中在别嘌呤醇不良反应密切相关的HLA-B*58:01位点上。对于汉族人群,Shuen-lu Hung等人的研究中纳入的51名别嘌呤醇-SCAR的患者中均存在HLA-B*58:01等位基因,但在对照组中只有17%的人携带此基因,则表明HLA-B*58:01等位基因是这种严重疾病的重要遗传风险因素[43]。在泰国人群中的研究则证明,HLA-B*58:01等位基因预测别嘌呤醇诱导的SJS/TEN的敏感性和特异性分别为100%和87%[44]。目前,对于HLA-B*58:01等位基因的检测已经用于临床痛风和高尿酸血症患者的指导用药并取得了良好的效果。
七、痛风的其他诊断指标
除了基因检测对痛风的诊断和治疗有着重要的临床价值,在既往的研究中发现临床常见的血常规和生化指标也与痛风有一定的相关性,如高尿酸的人群相较于正常人群嗜酸性粒细胞百分比、C反应蛋白、红细胞沉降率等指标会偏高;[24]而在高尿酸血症的患者中,HLA-B5801基因阳性的患者相较于阴性的患者嗜酸性粒细胞百分比和平均血小板体积会偏高。同时ALT/ALP比值、红细胞沉降率和中性粒细胞/淋巴细胞比值与尿酸的含量呈正相关[45]。这些指标的临床价值目前仍然未知,但是都有望在以后的痛风和高尿酸血症的诊断及别嘌呤醇药物效果预测中发挥重要作用。![]()
参考文献
Schlesinger N, Thiele RG.The pathogenesis of bone erosions in gouty arthritis. Ann Rheum Dis, 2010. 69(11): p. 1907-12.
Dalbeth N. Gout. The Lancet, 2021. 397(10287): p. 1843-1855.
Zhang X.Association between SLC2A9 (GLUT9) gene polymorphisms and gout susceptibility: an updated meta-analysis. Rheumatol Int, 2016. 36(8): p. 1157-65.
Zou, Y. Associations between the SLC22A12 gene and gout susceptibility: a meta-analysis. Clin Exp Rheumatol, 2018. 36(3): p. 442-447.
AkashiA. A common variant of LDL receptor related protein 2 (LRP2) gene is associated with gout susceptibility: a meta-analysis in a Japanese population. Hum Cell, 2020. 33(2): p. 303-307.
Hershfield MS. Clinical pharmacogenetics implementation consortium guidelines for human leukocyte antigen-B genotype and allopurinol dosing. Clin Pharmacol Ther, 2013. 93(2): p. 153-8.
Dalbeth N, Stamp L.Hyperuricaemia and gout: time for a new staging system? Ann Rheum Dis, 2014. 73(9): p. 1598-600.
王莉杰, 193例痛风患者的临床症状分析. 2015, 河南中医学院.
Shiozawa A. Serum uric acid and the risk of incident and recurrent gout: a systematic review. J Rheumatol, 2017. 44(3): p. 388-396.
Perez-Ruiz F. Clinical manifestations and diagnosis of gout. Rheum Dis Clin North Am, 2014. 40(2): p. 193-206.
Dalbeth N. Gout. Lancet, 2021. 397(10287): p. 1843-1855.
倪青. 高尿酸血症和痛风病证结合诊疗指南(2021-01-20). 世界中医药, 2021. 16(02): p. 183-189.
李林. 中国高尿酸血症相关疾病诊疗多学科专家共识. 中华内科杂志, 2017. 56(03): p. 235-248.
Zhu Y, Pandya BJ,Choi HK.Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum, 2011. 63(10): p. 3136-41.
Ragab G, Elshahaly M,Bardin T. Gout: An old disease in new perspective-A review. J Adv Res, 2017. 8(5): p. 495-511.
Kim JW. Prevalence and incidence of gout in Korea: data from the national health claims database 2007-2015. Rheumatol Int, 2017. 37(9): p. 1499-1506.
Zobbe K. Secular trends in the incidence and prevalence of gout in Denmark from 1995 to 2015: a nationwide register-based study. Rheumatology (Oxford), 2019. 58(5): p. 836-839.
Rai SK.The rising prevalence and incidence of gout in British Columbia, Canada: Population-based trends from 2000 to 2012. Semin Arthritis Rheum, 2017. 46(4): p. 451-456.
Robinson PC, Taylor WJ,Dalbeth N., An observational study of gout prevalence and quality of care in a national australian general practice population. J Rheumatol, 2015. 42(9): p. 1702-7.
Dehlin, M., L. Jacobsson, and E. Roddy, Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat Rev Rheumatol, 2020. 16(7): p. 380-390.
Choi HK. Purine-rich foods, dairy and protein intake, and the risk of gout in men. N Engl J Med, 2004. 350(11): p. 1093-103.
Choi HK. Alcohol intake and risk of incident gout in men: a prospective study. Lancet, 2004. 363(9417): p. 1277-81.
Choi HK, Curhan G.Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. Bmj, 2008. 336(7639): p. 309-12.
Fenando A. Gout, in statPearls. 2022, StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.: Treasure Island (FL).
Kolz M. meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet, 2009. 5(6): p. e1000504.
Köttgen A. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet, 2013. 45(2): p. 145-54.
Okada Y. meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat Genet, 2012. 44(8): p. 904-9.
Tin A. Genome-wide association study for serum urate concentrations and gout among African Americans identifies genomic risk loci and a novel URAT1 loss-of-function allele. Hum Mol Genet, 2011. 20(20): p. 4056-68.
Major TJ. An update on the genetics of hyperuricaemia and gout. Nat Rev Rheumatol, 2018. 14(6): p. 341-353.
Ekaratanawong S. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J Pharmacol Sci, 2004. 94(3): p. 297-304.
Bakhiya A. Human organic anion transporter 3 (hOAT3) can operate as an exchanger and mediate secretory urate flux. Cell Physiol Biochem, 2003. 13(5): p. 249-56.
Chiba T. NPT1/SLC17A1 is a renal urate exporter in humans and its common gain-of-function variant decreases the risk of renal underexcretion gout. Arthritis Rheumatol, 2015. 67(1): p. 281-7.
Van Aubel RA. Human organic anion transporter MRP4 (ABCC4) is an efflux pump for the purine end metabolite urate with multiple allosteric substrate binding sites. Am J Physiol Renal Physiol, 2005. 288(2): p. F327-33.
Bahn A. Identification of a new urate and high affinity nicotinate transporter, hOAT10 (SLC22A13). J Biol Chem, 2008. 283(24): p. 16332-41.
Eraly SA. Multiple organic anion transporters contribute to net renal excretion of uric acid. Physiol Genomics, 2008. 33(2): p. 180-92.
Vitart V. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet, 2008. 40(4): p. 437-42.
Enomoto A. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature, 2002. 417(6887): p. 447-52.
Woodward OM.Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc Natl Acad Sci U S A, 2009. 106(25): p. 10338-42.
Abhishek A, Roddy E,Doherty M.Gout-a guide for the general and acute physicians. Clin Med (Lond), 2017. 17(1): p. 54-59.
Zhang W. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis, 2006. 65(10): p. 1312-24.
陈光亮, 周媛凤, 张颖. 治疗痛风和高尿酸血症药物研究进展. 中国临床药理学与治疗学, 2017. 22(01): p. 104-109.
Bardin T, Richette P.The role of febuxostat in gout. Curr Opin Rheumatol, 2019. 31(2): p. 152-158.
Hung SI. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A, 2005. 102(11): p. 4134-9.
Tassaneeyakul W. Strong association between HLA-B*5801 and allopurinol-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogenet Genomics, 2009. 19(9): p. 704-9.
方忠俊. 痛风性关节炎患者HLA-B*5801基因型携带率及相关实验室指标分析. 检验医学, 2021. 36(09): p. 896-900.


