


宏基因组二代测序技术临床应用中假阳性及假阴性发生原因与防范措施
宏基因组二代测序技术(mNGS)是一项无偏倚的病原学诊断方法,可检测到样本中包括宿主和病原微生物的任何核酸信息。因此,mNGS在临床应用中带给临床医生更多的信息和诊断价值,同时也给医生带来一些疑惑,比如有些检测报告提示多种病原体,这导致临床很难甄别真正的致病原;而有些患者临床上明显存在感染征象,但mNGS结果却是阴性。那么,导致假阳性和假阴性的原因都有哪些,又该如何避免mNGS假阳性和假阴性的发生呢?本文就相关问题予以论述,同时给出防范措施。
一、mNGS假阳性发生的原因
1. 样本采集或运输流程中引入的微生物或核酸:如经皮穿刺,即使严格遵循无菌操作,皮肤表面或周围环境已死亡的微生物和核酸,仍有可能在采集过程中被引入,如表皮葡萄球菌、痤疮丙酸杆菌、棒状杆菌属及不动杆菌属等。
2. 正常人体定植微生物:微生物群落栖息在人体的各个部位,包括呼吸道粘膜、皮肤以及肠道等,这些菌群在维持人类健康方面发挥着重要作用。研究发现,健康人肺内也存在着连续多样的菌群生态系统[1]。此外,mNGS揭示了健康人外周血中也可能存在微生物核酸序列,这颠覆了人们原有的认知[2]。
3. 检测流程中的背景微生物:mNGS常检出较多背景微生物,包括试剂工程背景菌、环境微生物及实验室残留微生物。已有研究报道,背景微生物受所有实验室流程(包括样品预处理、核酸提取和文库制备)的影响,高水平的背景微生物并不代表检测性能差,可通过有效的数据过滤方式降低背景微生物所导致的假阳性结果[3]。

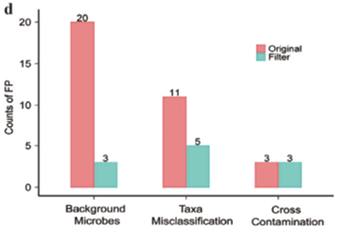
图1. 背景微生物受文库制备和核酸提取方法的影响(左)设置假阳性过滤器减少假阳性(右)
4. 样本间的交叉污染:样本间的交叉污染表示含有高丰度病原体的样本,交叉污染到其他样本,从而产生假阳性结果。样本的采集、运输、处理和实验过程中均有可能产生样本间的交叉污染。对于mNGS,交叉污染还可能是测序仪在混合测序时,高频标签(index)可能产生错误分配,导致标签跳跃(Index hopping)而引起。
5. 错误种属鉴定:微生物的分类依赖于参考数据库,使用非精密的数据库进行序列比对时,容易产生假阳性结果。研究者对参考数据库序列来源的评估发现,使用NCBI-NT数据库时,产生了多个虚假分类,导致较高的假阳性率和假阴性率[4]。在一些革兰氏阴性菌中的可移动元件,包括质粒、噬菌体、转座子、基因岛等,也会影响物种的鉴定。
此外,属内或种间相似性过高,数据库比对时,容易造成错分。如大肠埃希菌和志贺氏菌在基因组上相似度高,生信算法分辨率不足则可能造成错分。根据研究表明,实验室通过使用不同的算法,如Kraken、metaOthello以及其他自研生信算法等,可有效去除由于基因组同源性带来的交叉干扰[5]。
二、mNGS假阴性发生的原因
1. 送检样本质量:当样本质量不佳,选取非感染病灶或非原发病灶的标本,无责任病原微生物存在或含量低,则容易造成假阴性,如肺部感染或脑脓肿患者只检测外周血、脑脊液代替脑病的脑组织检测等。采样时间不合适是另一因素,如对急性病毒性脑膜炎患者,病毒只在发病最初几小时或几天存在于CNS,因此错过最佳时间采集脑脊液样本可能无助于mNGS检测。此外,保存和运输条件不当可能导致微生物核酸(尤其是RNA)的降解,从而导致结果阴性。
2. 人源核酸含量过高:高水平的人源核酸会影响mNGS对病原微生物检测的灵敏度,因为人基因组约比细菌基因组大1000倍(~3×109 bp vs ~3×106 bp),人源核酸可以占样本测序序列的95%以上[7]。
3. 核酸提取效率低:某些病原体(如真菌、结核分枝杆菌)的细胞壁较厚,核酸提取困难,而对于无细胞(如病毒)或细胞壁较薄的病原微生物,即使应用常规的破壁方法,也会造成核酸损失,两种情况均会造成假阴性的结果。
4. 数据库不完善:微生物数据库中,如罕见或新发病原体菌株参考序列不完整、参考数据库具有物种偏好性以及遗传相似性过高的种属出现错误分类等,均可能导致假阴性结果。
此外,检测项目选择不当、测序数据量不足,均可能造成与临床诊断不相符的情况。
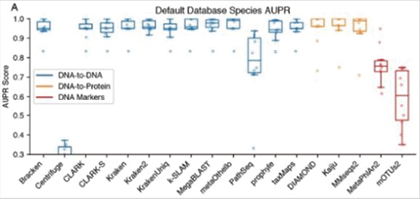
图2. 不同分类算法对物种级别鉴定的准确性评估[6]
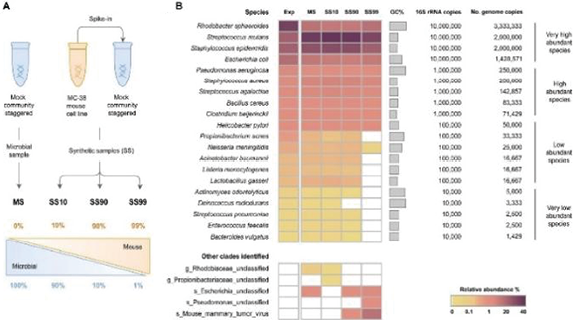
图3. 当人源核酸序列数占比达97%~99%时,病原微生物检测灵敏度显著降低[8]
三、假阳性与假阴性防范措施
1. 合格的样本是检测结果质量的保证:严格无菌操作,当采集到病灶处标本后立即送检,避免耽搁时间过长。
2. 提高核酸提取效率:不同微生物细胞结构差异大,提取流程中破壁过程和条件均影响核酸提取效率。研究发现珠磨破壁可以不同程度的提高9种常见细菌、真菌的检出[9],然而珠磨破壁会降低RNA病毒的检出[3]。因此采用双流程、“双重破壁”方式,不仅可以提高目标病原体的序列数,同时减少RNA病毒损失。
3. 选择性去人源:对于人源细胞浓度高的样本(如胸腔积液、严重血性脑脊液等),为提高低载量病原体的检出率,添加去人源过程。
4. 构建完整且高质量的病原微生物数据库:对权威数据库中基因组序列进行挑选、整理、分类,过滤低质量基因组,去除冗余,通过程序软件将收集到的基因组序列整理成病原微生物和人源序列比对数据库。并保持更新。
5. 建立试剂及环境背景核酸数据库:mNGS检测过程中试剂背景菌核酸片段可导致结果假阳性;一些样本(如呼吸道样本)会存在定植微生物,因此,需要构建背景库用于过滤污染序列、区分背景病原体和致病性病原体。
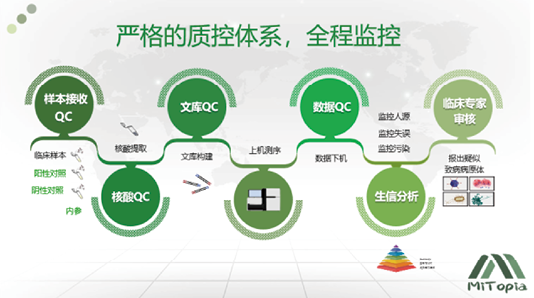
6. 全流程质控体系:每批实验需设置阴性对照品和阳性对照品,并在标本中加入内标,在核酸提取、文库构建环节分别设置质控点,同时对下机数据进行全面的质控。
临床研究表明,mNGS检出的病原微生物即标本中真实存在的病原微生物,有可能是致病微生物、定植微生物或者污染菌,这些都是检测的真阳性,只是不一定都致病。而如何判断是否是致病微生物,需要提高mNGS检测准确性和灵敏度,降低mNGS报告的假阴性率以排除感染,同时尽可能减少污染,避免假阳性对临床判断造成的干扰。
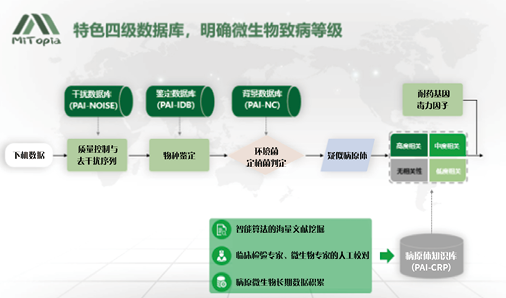
欧蒙未一医学检验实验室(以下简称“未一医学”)成立于2014年,是国内最早一批开展病原微生物mNGS检测的第三方实验室。未一医学已在北京、杭州和广州分别设有独立医学检验实验室,致力于为风湿免疫科、呼吸科、神经科、儿科等领域提供“多平台”、“一体化”疾病诊断解决方案,覆盖疾病类别逾两千种,合作的三甲医院超千家。质量是立足的根本,创新是前进的动力。未一医学秉承认真、严谨的精神,不断创新,积极推动先进诊断技术在临床和科研领域的应用与发展。
参考文献
Man WH, de Steenhuijsen Piters WA, Bogaert D. The microbiota of the respiratory tract: gatekeeper to respiratory health[J]. Nat Rev Microbiol, 2017, 15(5): 259-270.
Li N, Cai Q, Miao Q, et al. High‐Throughput metagenomics for Identification of Pathogens in the Clinical Settings[J]. Small Methods, 2020.
Liu D, Zhou H, Xu T, et al. Multicenter assessment of shotgun metagenomics for pathogen detection[J]. EBioMedicine, 2021, 74: 103649.
van Boheemen S, van Rijn AL, Pappas N, et al. Retrospective Validation of a metagenomic Sequencing Protocol for Combined Detection of RNA and DNA Viruses Using Respiratory Samples from Pediatric Patients[J]. J Mol Diagn, 2020, 22(2): 196-207.
Jing C, Chen H, Liang Y, et al. Clinical evaluation of an Improved metagenomic Next-Generation Sequencing Test for the Diagnosis of Bloodstream Infections[J]. Clin Chem, 2021, 67(8): 1133-1143.
Ye SH, Siddle KJ, Park DJ, et al. Benchmarking metagenomics Tools for Taxonomic Classification[J]. Cell, 2019, 178(4): 779-794.
Marotz CA, Sanders JG, Zuniga C, et al. Improving saliva shotgun metagenomics by chemical host DNA depletion[J]. Microbiome, 2018, 6(1): 42.
Pereira-Marques J, Hout A, Ferreira RM, et al. Impact of Host DNA and Sequencing Depth on the Taxonomic Resolution of Whole metagenome Sequencing for Microbiome Analysis[J]. Frontiers in microbiology, 2019, 10: 1277-1277.
Schlaberg R, et al. Validation of metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection[J]. Arch Pathol Lab Med, 2017, 141(6): 776-786.


