


血友病患者基因诊断策略与存在的问题

戴菁,博士,主任技师,检验医师,博士生导师。长期从事血栓性和出血性疾病的临床和分子发病机制研究,作为主持人承担国自然科学基金面上项目,参与科技部重点项目课题研究等,发表SCI论文十余篇,作为主要完成人参与的科研项目先后获得上海市科技进步奖一等奖、中华医学科技奖二等奖、教育部科技二等奖等。主要学术兼职:中国研究型医院学会血栓与止血专业委员会常务委员,中国医师协会检验医师分会青年委员,上海市医学会检验分会青年学组成员,上海市中西医结合学会第二届检验医学专业委员会委员,中国老年医学学会检验医学分会委员。

王学锋,主任医师,二级教授,博士生导师。现任上海交通大学医学院医学技术学院副院长、检验系主任,附属瑞金医院实验诊断中心主任、检验科主任,临床输血科主任;上海市领军人才。主要学术兼职:现任上海市医学会检验医学分会候任主委,全国临床输血委员会副主任,中华医学会检验分会血液学和体液学学组副组长。获国家科技进步奖3项,省部级科技奖10余项;对出血性疾病和血栓性疾病建立了完善的实验室诊断体系,使这两类疾病的临床诊断水平与国际接轨。
血友病是常见的遗传性出血性疾病,分为A(hemophilia A,HA)和B(hemophilia B,HB)两型,其发病原因是凝血因子Ⅷ基因(F8)或Ⅸ基因(F9)缺陷导致蛋白的合成缺陷或质的异常,或者两者兼有之。根据国际血友病联盟(World Federation of Hemophilia,WFH)的统计,男性HA发生率为1/5000,HB则约为HA的1/5,且疾病的发生与地域、种族无关[1]。我国人口众多,血友病患者的绝对人数相当可观,据中国血友病协作组统计[2],自我国开展的血友病患者信息登记工作以来,目前共登记的血友病患者HA人数为23000人,HB人数为3600人。近年来,随着实验室筛选实验和检测方法的不断改进,对轻型血友病患者的诊断率提高,发病率有所上升。
一、血友病的发病机制
F8基因位于Xq28,全长186Kb,由26个外显子和25个内含子组成,FⅧmRNA全长约9Kb,编码了由2351个氨基酸残基(Amino Acid,AA)组成的多肽链,其中包括一条19个氨基酸组成的信号肽,成熟的肽链由2332个氨基酸以A1-A2-B-A3-C1-C2的方式排列,FⅧ蛋白分泌入血后,在血液中与血管性血友病因子(vonWillebrand Factor,vWF)结合,以FⅧ/vWF复合物形式存在,后者可稳定FⅧ,防止其过早降解。F8基因不仅结构庞大,而且导致HA的基因突变种类繁多,其中基因内含子22倒位突变引起的FⅧ蛋白缺乏,占重型血友病A分子发病机制的42%[3];而内含子1倒位的发生率大约占重型血友病A的2~5%[4],其余几乎每个家系都有不同的突变。目前,F8基因突变数据库报道的有2015种各种不同的突变[5],包括点突变、基因缺失、插入、无义突变、剪接突变等,从而提示F8基因异常具有高度的异质性;中型和轻型血友病中,86%为错义突变。上海瑞金医院分别对933例有家族史的HA患者以及393例散发HA患者基因突变谱进行整理分析(图1),各突变类型与突变数据库的报道结果类似[6]。
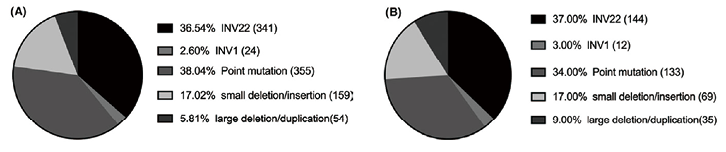
图1:A为933例有家族史的HA患者基因突变谱;B为393例散发HA患者基因突变谱
F9基因片段较小,相对于F8而言结构较为简单,长约为34kb,含8个外显子,FⅨ mRNA长约2.8kb,编码461个氨基酸的多肽链前体,经信号肽酶和蛋白酶切除信号肽和原肽,并经过糖基化、二硫键形成、氨基端12个氨基酸羧基化及第I类表皮生长因子区第64位天冬氨酸β羟化等一系列化学修饰后形成含415个氨基酸的成熟的FⅨ。FⅨ基因突变包括缺失、插入和点突变,目前F9突变数据库收录的突变位点有1094种,其中约80%为单个碱基突变[7]。
二、血友病基因诊断
通过基因诊断可以更精准地为血友病患者及其家属开展优生优育指导,尤其是对家族中有生育希望及可能的女性进行携带者基因检测,对优生优育有重要的价值[8-11]。同时,对血友病患者进行明确的基因诊断有利于接受预防治疗的患者更明确地知晓导致疾病的基因类型,越来越多的研究提示某些特殊的基因突变位点和抑制物的发生相关[12-14],针对不同突变位点采取不同治疗方案的措施也在逐步的探索中,有望在未来实现血友病患者的个体化治疗[15]。我们就目前常用的基因诊断的策略及基因诊断存在的问题做一简要评述。
1. 血友病A基因诊断常规流程:(1)内含子倒位的检测:内含子22倒位是20%HA患者的致病突变,是45%重型HA患者的致病突变。内含子22倒位是F8基因第22内含子内的Int22h序列与基因外的两个与之高度同源的序列(Int22h-2,Int2h-3)之一发生染色体内的同源重组(图2右),导致FⅧ基因在外显子1~22与23~26间发生断裂,不能合成正常的凝血因子FⅧ,导致重型血友病A。内含子1倒位是继内含子22倒位后被发现的另一导致血友病A的重要发病机制,系内含子1内的Inth-1与基因外的高度同源的序列Inth-2发生染色体内的同源重组(图2左),导致FⅧ蛋白不能正常合成。因此,首先需要对HA患者的F8基因内含子22倒位和内含子1倒位进行检测。经典的用于检测内含子22倒位的方法是Southern Blot法,但该方法费时费力,并且需要使用荧光标记物品,不利于实验室开展[16];长链PCR(LD-PCR)方法是由Steve Sommer设计的一种简便的检测内含子22倒位的检测方法,但由于需要扩增的片段长度分别为11kb和12kb,因此该检测方法对技术要求比较高[17];Rossetti等用Inverse shifting-PCR(IS-PCR)的方法进行内含子22倒位的检测,虽然可将扩增片段缩小,但由于其要求大量的DNA(1-2μg)以及很多步骤的酶切、自连接过程,使用起来也不是很方便[18];本课题组建立了一种结合LD-PCR和AccuCopy技术定量的方法对内含子22倒位进行检测[19],能准确地区分各种不同类型的倒位组合,使得内含子22倒位检测的便利性和准确性得到了极大的提升。当内含子22倒位或者内含子1倒位检测结果为阴性时,我们需要对F8基因的编码序列进行检测。
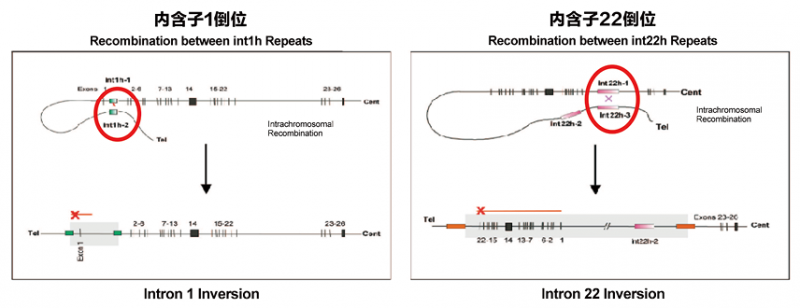
图2:内含子1倒位和22倒位示意图
(2)F8基因26个外显子及其侧翼序列的基因检测:分别针对F8基因的共26个外显子以及5’端和3’端非编码区设计特异性引物,运用PCR的方法将上述目的片段进行扩增,随后采用Sanger法进行基因序列的检测。使用该方法可以检测出绝大部分的F8基因突变位点,包括小片段的缺失、插入、点突变(无义突变和错义突变)、剪切位点突变等。F8基因的第14号外显子是最大的外显子片段,该外显子编码FⅧ蛋白的B区,约占总编码区的43%,有研究[20]显示21%的点突变可发生在该外显子区段,但更多的是小片段的缺失和插入(约占47%),虽然B区在FⅧ蛋白成熟过程中会被剪切,但发生在该区段的基因改变仍可影响FⅧ蛋白的分泌效率[21, 22]。
(3)间接基因诊断:对于HA,由于F8基因不仅庞大且结构复杂,直接查找突变费时费力,故可以采用结合遗传连锁分析的间接诊断方法进行血友病A的携带者与产前诊断,发掘高信息量的多态性位点并通过遗传连锁分析的方法可以定位致病基因所在的染色体,但这些多态性位点的信息量在各个人群中存在差异性,如在欧美国家有高诊断信息量的常用位点(STR、CA12、CA22、BclI)在国人的信息量仅为27%[23],我们先后开发了适合中国人群的二代、三代连锁分析位点结合性别位点,利用多重PCR一次扩增,累计识别能力达到了99%以上[24],很好地弥补了直接诊断的短板,但由于基因的复杂性,遗传连锁分析所选择的位点并不能完全避免基因重组的发生对结果的干扰和误判。
2. 血友病B基因诊断流程:由于FⅨ基因较小,可以采用直接的核酸测序方法进行基因突变检测;F9基因外的6个多态性位点经过多重PCR扩增可用于血友病B的遗传连锁分析,但也无法排除基因重组的发生对其结果的干扰。
3. 特殊血友病患者的基因检测方法:(1)拷贝数检测:运用上述基因诊断策略基本能对90%以上的HA患者做出明确的基因诊断,但仍有10%左右的患者未能找到明确的突变位点,需要用其他的技术进行诊断。基因拷贝数变化也是导致蛋白异常表达的一种基因改变,但用常规的测序技术并不能检出,多重连接依赖式探针扩增技术(multiplex ligation-dependent probe amplication,MLPA)可用于检测基因拷贝数的变化,如双重拷贝、三重拷贝等异常[25, 26]。AccuCopy是一种在多重竞争性PCR扩增的基础上进行拷贝数变异性检测的技术[27],利用该技术我们可以对目的基因片段中的拷贝数情况进行检测,我们对10例利用常规方法未找到基因异常位点的HA患者进行了AccuCopy拷贝数检测[28],均明确了拷贝数异常的位点,为HA基因诊断提供了可靠的补充检测手段。
(2)NGS(Next-Generation Sequencing)检测:NGS又称为二代测序[29],这是相对于Sanger一代测序而言,是近几年发展起来的一种高效率的基因测序方法,其检测深度和广度、高通量等特性使得其在临床上得到了广泛应用,比如肿瘤等。Sanger法测序其需要事先知道目的基因的序列,但NGS则没有这个要求,从而使得NGS方法可以发现更多未知的致病基因,例如在儿科,多种先天性疾病之前没办法明确致病基因,但随着NGS方法的运用,越来越多的先天性疾病得到了明确的基因诊断。由于NGS方法获得的数据量信息很大,如何从海量的信息中捕获具有疾病诊断意义的异常基因需要经验丰富的科研人员进行筛选。Jose Maria Bastida等[30]建立了一种针对F8、F9和VWF基因的NGS检测组合,对102份标本(97例HA/HB患者和5例女性携带者)进行了检测,其诊断率可达到99%;应用NGS方法对F8基因进行检测还可以发现内含子深部的位点变异,这种类型的变异往往可导致轻型或中型血友病的发生,这也是为什么轻型或中型血友病用常规基因检测方法未能找到基因异常的原因。
(3)女性HA的基因诊断:女性HA十分罕见,首先需要注意和血管性血友病(von Willerbrand Disease,VWD)在表型上进行鉴别;在对F8基因进行检测后,若能发现基因缺陷,则对确立女性血友病的诊断具有重要价值。上海瑞金医院诊断的3例女性血友病A患者中的2例分别是内含子22倒位和内含子1倒位导致的重型血友病A,其另一条X染色体非随机灭活;另一例女性患者存在双杂合突变导致重型血友病A的发生[31]。
(4)散发家系:根据文献报道,大约1/3的血友病A家系为没有家族史的散发家系,对于这些家系,目前主要通过直接诊断和间接诊断相结合的策略。但散发家系的诊断需要考虑细胞嵌合现象的存在,本课题组[6]对2005年至2016年的十年间来院就诊的393个散发HA家系的基因突变进行分析发现,大部分散发血友病A患者的母亲为携带者,其致病基因的来源可追溯到患者的外公(60%)或者母亲(12%),28%的散发患者为自发突变,体细胞嵌合现象在散发家系中存在的比例较高。
三、血友病基因诊断存在的问题
由于基因诊断是近十几年来逐渐在临床上推广应用的新技术,对于疾病与基因之间的关系也随着越来越多的研究工作的开展而逐渐揭开了神秘的面纱,我们所面临的问题也越来越清晰、明确。
1. 检测技术问题:以F8基因内含子22倒位(Int22)检测为例,从最早发明该突变热点的检测技术以来,检测方法不断更新换代,从Southen Blot、LD-PCR、IS-PCR到AQ-PLP等技术,使得对Int22的检测方法灵敏度和准确度都得到了很好地改善,各种非经典的Int22倒位类型不断的被发现,细胞嵌合标本的最低检测率可达到2%,有效地提高了散发家系基因突变的检出率。我们利用AQ-PLP技术对一例无家族史的HA家系进行Int22检测[32],发生该先症者F8基因存在一种新型的22倒位模式(int22h-2、int22h-3、int22h-2/-1),并通过对其母亲的各种体细胞嵌合率检测结果分析认为其母亲为嵌合携带者,且突变发生在胚胎发育早期。
FⅧ和FⅨ基因结构异常也是影响基因检测的一个重要因素,常规技术对基因结构异常无法检出,据统计,在重型HA和HB患者中存在结构异常的概率分别为6%和10%[33]。结构异常合并突变位点同时存在的案例也有见报道[34],进一步体现了血友病基因诊断的复杂性。
2. 患者家族对基因诊断的认识:据2012年美国的一项针对血友病基因检测的患者调查结果显示[35, 36],仅有20%的血友病患者进行了基因检测,分析其原因,一部分是由于检测费用高昂,无法由保险支付;另一个重要原因就是缺乏可靠的检测技术手段。目前所有的检测技术由于各种原因限制以及基因突变复杂性、异质性等特点的影响,均无法达到100%的检测准确率,加拿大的一项对十年间全国血友病基因检测的研究数据表明,HA和HB的基因检测可诊断率分别为91%和94%[20];随着检测技术的不断发展,即使是最新的NGS技术用于血友病的基因检测,仍有患者无法得到明确的基因结果[37, 38],利用最新的NGS技术结合MIP分子检测策略,HA和HB的可诊断率分别达到了98.1%和99.3%[39],但仍然有血友病患者在所有能使用的基因检测方法后仍无法明确基因异常位点。
综上所述,血友病基因检测所面临的问题和挑战并存,对于血友病患者来说,他们需要正确认识血友病基因诊断在疾病的治疗、优生优育以及抑制物产生的指导作用,以及基因诊断的现状;医务科研工作者需要不断地改进基因检测技术以及分析基因诊断结果与患者临床表现的相关性,梳理基因与疾病的关系,更好地为血友病的基因治疗、个体化治疗探索基因宝库。
参考文献:
Keeney S, Mitchell M, Goodeve A, et al. The molecular analysis of haemophilia A: a guideline from the UK haemophilia centre doctors' organization haemophilia genetics laboratory network. Haemophilia. 2005 Jul; 11(4): 387-97.
中华医学会血液学分会血栓与止血学组,中国血友病协作组. 血友病诊断与治疗中国专家共识(2017年版). 中华血液学杂志, 2017, 38(5): 264-370
D. Lakich, H.H. Kazazian Jr., S.E. Antonarakis, et al. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A, Nat. Genet, 1993; 5(3): 236-241.
Bagnall RD, Waseem N, Green PM, et al. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood, 2002; 99(1): 168-174
王学锋, 吴竞生, 胡豫等, 临床出血与血栓性疾病, 人民卫生出版社, 2018
Y Lu, Y Xin, J Dai, X Wu, G You, Q Ding, W Wu, X Wang*. Spectrum and origin of mutations in sporadic cases of haemophilia A in China. Haemophilia, 2018; 24(2): 291-298
Lu Y, Wu X, Dai J, Ding Q, Wu W, Wang X. The characteristics and spectrum of F9 mutations in Chinese sporadic haemophilia B pedigrees. Haemophilia, 2019; 25(2): 316-323
Chalmers E, Williams M, Brennand J, Liesner R, Collins P, Richards M; Paediatric Working Party of United Kingdom Haemophilia Doctors’Organization. Guideline on the management of haemophilia in the fetus and neonate. Br J Haematol. 2011; 154(2): 208-215.
Gouw SC, van den Berg HM, Oldenburg J, et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: systematic review and meta-analysis. Blood. 2012; 119(12): 2922-2934.
Carcao MD, van den Berg HM, Ljung R, Mancuso ME. Correlation between phenotype and genotype in a large unselected cohort of children with severe hemophilia A. Blood. 2013; 121(19): 3946-3952.
Swystun LL, James P. Using genetic diagnostics in hemophilia and von Willebrand disease. Hematology Am Soc Hematol Educ Program. 2015; 2015: 152-159
Oldenburg J, Pavlova A. Genetic risk factors for inhibitors to factors VIII and IX. Haemophilia. 2006; 12 (suppl 6): 15-22.
DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br J Haematol. 2007; 138(3): 305-315.
Eckhardt CL, van Velzen AS, Peters M, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood, 2013; 122(11): 1954-1962
James PD, Raut S, Rivard GE, et al. Aminoglycoside suppression of nonsense mutations in severe hemophilia. Blood. 2005; 106(9): 3043-3048.
Naylor JA, Buck D, Green P. et al. Investigation of the factor VIII intron 22 repeated region (int22h) and the associated inversion junctions. Hum Mol Genet, 1995; 4(7): 1217-1224
Liu Q, Thorland EC, Heit JA, et al. Overlapping PCR for bidirectional PCR amplification of specific alleles: a rapid one-tube method for simultaneously differentiating homozygotes and heterozygotes, Genome Res, 1997; 7(4): 389-398
Rossetti LC, Radic CP, Larripa IB, et al. Genotyping the hemophilia inversion hotspot by use of inverse PCR, Clin Chem, 2005; 51(7): 1154-1158
Qiulan Ding, Xi Wu, Yeling Lu, et al. AccuCopy quantification combined with pre-amplification of long-distance PCR for fast analysis of intron 22 inversion in haemophilia A. Clin Chim Acta, 2016; 458: 78-83
Rydz Natalia, Leggo Jayne,Tinlin Shawn, et al. The Canadian“National Program for hemophilia mutation testing”database: A ten-year review. Am. J. Hematol, 2013; 88: 1030-1034,
Pittman DD, Alderman EM, Tomkinson KN, et al. Biochemical, immunological,and in vivo functional characterization of B-domaindeleted factor VIII. Blood, 1993; 81: 2925-2935.
Miao HZ, Sirachainan N, Palmer L,et al. Bioengineering of coagulation factor VIII for improved secretion. Blood, 2004; 103: 3412-3419.
王学锋, 刘元昉, 李广志等, 血友病A携带者检测与产前诊断. 中华血液学杂志, 2001, 22: 117-120
Ding QL, Lu YL, Dai J, et al. Characterisation and validation of a novel panel of the six short tandem repeats for genetic counselling in Chinese haemophilia A pedigrees. Haemophilia. 2012; 18(4): 621-625.
Rost S, L€offler S, Pavlova a, et al. Detection of large duplications within the factor VIII gene by MLPA. J Thromb Haemost 2008; 6: 1996-1999.
Acquila M, Pasino M, Di Duca M, et al. MLPA assay in F8 gene mutation screening. Haemophilia 2008; 14: 625-627.
R. Du, C. Lu, Z. Jiang, et al. Efficient typing of copy number variations in a segmental duplication-mediated rearrangement hotspot using multiplex competitive amplification. J. Hum. Genet, 2012; 57(8): 545-551.
You GL, Ding QL, Lu YL, et al. Characterization of large deletions in the F8 gene using multiple competitive amplification and the genome walking technique. J Thromb Haemost, 2013; 11(6): 1103-1110.
Simon AH, Ira WD, Tim RM. Reference standards for next-generation sequencing. Nature Review, 2017; 18(8): 473-484.
Jose MB, Jose RG, Cristina J, et al. Application of molecular diagnostic algorithm for haemophilia A and B using next-generation sequencing of entire F8, F9 and VWF genes. Thromb Haemost, 2017; 117(1): 66-74.
Xuefeng Wang, Yeling Lu, Qiulan Ding, et al. Haemophilia A in two unrelated females due to F8 gene inversions combined with skewe32inactivation of X chromosome. Thromb Haemost 2009; 101(4): 775-778.
Chen C, Xie X, Wu X, Lu Y, Wang X, Wu W, Hu Y, Ding Q. Complex recombination with deletion in the F8 and duplication in the TMLHE mediated by int22h copies during early embryogenesis. Thromb Haemost. 2017; 117(8): 1478-1485.
Miller CH, Benson J, Ellingsen D, et al; Hemophilia Inhibitor Research Study Investigators. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. 2012; 18(3): 375-382.
Shetty S, Bhave M, Ghosh K. Challenges of multiple mutations in individual patients with haemophilia. Eur J Haematol. 2011; 86(3): 185-190.
Centers for Disease Control and Prevention (CDC). CDC Hemophilia A Mutation Project (CHAMP). Available at: http://www.cdc.gov/ncbddd/hemophilia/champs.html. Accessed March 2016.
Centers for Disease Control and Prevention (CDC). CDC Hemophilia B Mutation Project (CHBMP). Available at: http://www.cdc.gov/ncbddd/hemophilia/champs.html. Accessed March 2016.
Simeoni I, Stephens JC, Hu F, et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood. 2016; 127(23): 2791-2803.
Bastida JM, Del Rey M, Lozano ML, et al. Design and application of a 23-gene panel by next-generation sequencing for inherited coagulation bleeding disorders. Haemophilia. 2016; 22(4): 590-597.
Jill M. Johnsen,Shelley N. Fletcher, Haley Huston,et al. Novel approach to genetic analysis and results in 3000 hemophilia patients enrolled in the My Life, Our Future initiative. Blood advances, 2017; 13(1): 824-834


