


同位素稀释液相色谱串联质谱法检测血清C肽候选参考方法的建立与性能验证

邓宇航,北京协和医学院临床检验诊断学在读博士(培养单位:国家卫生健康委临床检验中心),国家留学基金委公派新加坡国立大学联合培养博士。以第一作者或共同第一作者发表SCI论文7篇,主要研究方向为生化检验参考体系的建立与应用。从事质谱参考方法研究及应用四年,完成质谱类参考方法建立2项。
【摘要】目的 基于同位素稀释液相色谱串联质谱技术,建立人血清C肽测定的候选参考方法。方法 准确称量的血清和校准溶液在与C肽同位素内标混合后,加入硫酸锌溶液沉淀蛋白质,对离心后的上清液进行阴离子交换固相萃取,提纯C肽和内标,最后氮气吹干复溶,用LC/MS/MS定量检测C肽。基于人血清样本,评价该方法的精密度、加标回收率和测量不确定度等;并分别以信噪比(S/N)≥3和S/N≥10作为标准,评价方法的检测限和定量限。ID-LC-MS/MS法与2种常规方法同时检测76份单人份血清样本,并对检测结果进行普通线性回归分析和差异分析。结果 测定4个浓度水平混合血清5个批次,方法的批内、批间和总变异系数(CV)的均值(范围)分别为1.0%~2.1%,0.6%~1.2%,和1.3%~2.2%。低中高3个浓度水平样本的加标回收率的均值(范围)分别为100.3%~100.7%,100.4% ~101.0%,和99.6%~100.7%。方法的定量限和检测限分别为33.3和10.0pmol/L,并与两种常规方法有强相关性(R2 > 0.98)。结论 建立了一种新的ID-LC/MS/MS方法测定血清C肽,该方法精密,准确,有望作为血清C肽测定的参考方法。
【关键词】血清C肽;液相色谱串联质谱;方法对比;糖尿病;标准化
糖尿病是人类最常见的慢性代谢性疾病之一,其具有全身性进行性发展,并发症多且严重等特点。目前,随着生产的发展、生活水平的提高及人口寿命的延长, 糖尿病的发病率逐年增长,已成为世界各国越来越严重的公共卫生难题,给社会带来了严重的健康问题和巨大的经济负担[1, 2]。内源性胰岛素分泌量检测及胰岛β细胞分泌功能检测对糖尿病患者的分型和治疗监测至关重要[3, 4]。C肽是由31个氨基酸组成的多肽,它与胰岛呈等分子量分泌,因此常被用来评估糖尿病患者胰岛β细胞分泌内源性胰岛素的能力[5]。相对于直接测量胰岛素,C肽检测往往更能准确反应胰岛β细胞的分泌功能,首先体内C肽较胰岛素有更长的半衰期,其在血液中的浓度是胰岛素的3到5倍,因此,其体内浓度波动较胰岛素小,并有着更稳定的检测窗口[6, 7]。其次,由于一型糖尿病患者以及晚期二型糖尿病患者往往需要注射外源性胰岛素进行治疗,这些外源性胰岛素易对胰岛素检测造成干扰,而C肽检测则不受影响,因此,这时候C肽检测则更能准确的反应内源性胰岛素的分泌情况。
目前我国临床实验室多采用商业化的免疫法来定量血清C肽。然而,我们之前的研究表明,不同实验室间的测量结果存在明显的差异,不具有可比性[8]。因此,亟需参考方法来改进常规检测的正确性,以利于提高实验室之间检测结果的一致性,改善C肽检测的标准化现状。同位素稀释液相色谱串联质谱法(ID-LC-MS/MS)特异、准确且灵敏,被广泛用作(包括C肽)众多临床指标的参考方法[9, 10]。为了提高血清C肽检测实验室间的一致性,本研究基于ID-LC-MS/MS技术,建立了血清C肽检测的候选参考方法,该方法的性能得到了充分的验证,并与两种广泛使用的常规免疫方法进行了对比。
材料与方法
一、仪器和试剂及血清样本
1.仪器:LC-MS/MS系统由Waters Acquity UPLC Fl-I型高效液相色谱仪(Waters,美国)和API6500 TripleQuadruple质谱仪(ABSCIEX,美国)组成。 Milli-Q纯水机(Merck,德国),十万分之一天平(Max=120g,d=0.01mg,Sartorious,德国),台式高速冷冻离心机(Sigma 3k15,德国),氮吹仪(Thermo,美国)。两种常见常规免疫检测系统:Dxi800(Beckman,美国),ADVIA Centraur XP(Siemens,德国),均使用其配套的校准品和试剂。
2.化学试剂:标准品为国际检验医学溯源联合委员会(JCTLM)收录的一级参考物质NMIJ CRM 6901-c,购自于日本国家计量院。内标为碳13标记的C肽(27,31-13C-peptide),由上海吉尔生化合成。色谱纯乙腈和甲醇购自美国Thermo Fisher Scientific公司。甲酸和硫酸锌购自德国Sigma-Aldrich公司。
3.样本:精密度评价样本为混合人血清,收集自北京医院内分泌实验室;评价回收率的基础样本为浓度1.86ng/g的新鲜混合血清。方法对比样本为76份单人份血清样品,均收集自北京医院内分泌实验室和检验科。以上样本在使用前均保存在-80℃。
二、标准溶液和内标溶液的制备
根据使用说明,首先将标准品(NMIJ CRM 6901-c)配置成104±5mg/L的一级标准溶液,随后用胎牛血清对一级标准溶液进行逐级稀释,制备成898.3ng/g的储备液和10.4ng/g的工作液。与此类似,将同位素标记C肽溶解在胎牛血清中并逐级稀释,最终制备浓度为27.5ng/g的内标工作液。所有配置过程均严格采用重量法,完成后分装成1mL一瓶,储存于-80℃。
三、定量方法
本研究采用包括法定量,适量的血清样品和标准溶液中加入一定量的内标溶液,精确记录样本、标准溶液和内标液的质量,保证血清样品、低浓度标准溶液和高浓度标准溶液中C肽与内标的峰面积比分别为1.0、0.9和1.1。计算公式如下:
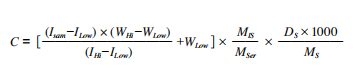
其中C为血清C肽浓度(pmol/L),ISam、ILow和IHi分别为血清样品、低浓度标准溶液和高浓度标准溶液中C肽和内标的峰面积比;WLow和WHi分别为低浓度和高浓度标准溶液的C肽和内标质量比;MIS是血清样品中内标的质量;MSer和DS分别为血清样品的质量和密度;MS为C肽的相对分子质量。
四、样本前处理
1. 标准液与内标液的混合:分析每批血清样本需制备一对标准。用微量移液器取50μL内标液和适量标准液,精确记录加入的内标液和标准液的质量,轻轻振荡混匀。该混合液中C肽与内标的峰面积之比约为0.9,记为低标。按上述方法取适量标准液与50μL内标液混合,该混合液中C肽与内标的峰面积之比约为1.1,记为高标。
2. 血清样本与内标的混合:常规方法对待测样本进行初测,得到C肽的大概浓度。用微量移液器取适量(0.05~1.0mL)血清与50μL内标液混合,并精确记录标准溶液与内标溶液的质量,使混合液中C肽与内标的峰面积比近似为1.0。加入适量胎牛血清,使终体积为1.0mL,轻轻混匀。
3. C肽提取:向混合液中加入1mL ZnSO4溶液(0.1mmol/L,pH=3.5~4.0),涡旋以沉淀蛋白质,离心10分钟(21100g,4℃),转移上清液至甲醇和水活化后的混合阴离子交换(Oasis MAX,Waters,美国)柱中,用1mL ZnSO4溶液(0.1mmol/L,pH=3.5~4.0)和1mL甲醇依次淋洗。最后,用2mL含1%甲酸的甲醇进行洗脱,氮气吹干后,用100μL含0.4%甲酸的乙腈/水溶液(1:9)复溶。
五、LC-MS/MS分析
1. 色谱条件:液相色谱柱为XSelect Peptide CSH C18柱(3.5μm,2.1×100mm,Waters,美国),柱温为40℃。流动相A为0.4%甲酸水,流动相B为0.4%甲酸乙腈,流速为0.4mL/min,单次检测时间为8分钟。梯度洗脱程序为:0分钟,10% B,2分钟,25% B;3.5分钟,40% B;5.5分钟,40% B;5.51分钟,100% B;6.5分钟,100% B;6.51分钟,10% B;8分钟,10% B。
2. 质谱条件:质谱仪采用多反应监测模式(Multiple reaction monitor,MRM)和正电喷雾电离模式进行电离(ESI+)。C肽母子离子质荷比(m/z)分别为1007.7(3+)和147.2(1+);内标母子离子m/z分别为1011.7(3+),147.2(1+)。质谱仪相关参数为:源气体温度:400℃;离子喷雾电压:5500V;气帘气(CUR):30psi;雾化器气体(GS1):90psi;辅助气体(GS2):60psi;碰撞气体(CAD):8psi;去簇电位(DP):120V;入口电位(EP):7V;碰撞能量(CE):28eV;碰撞出口电位(CXP):12V;Q1分辨率:低(Low),Q2分辨率:中(Unit)。血清样品中C肽和内标的LC-MS/MS图见图1。
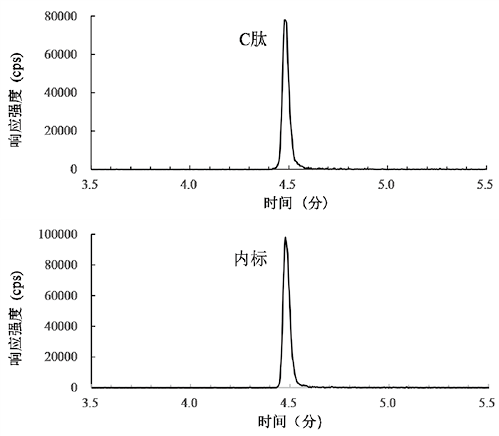
图1. 血清样本中C肽与内标的液相色谱串联质谱图
六、方法性能评价
1.基质效应、样品前处理绝对回收率、定量限(LOQ)和检测限(LOD):首先向血清样本中加入一定量的内标,然后进行前处理;另一组样本则对血清样本先直接进行前处理,再向前处理后的基质中加入等量的内标;最后对两份样本进行LC-MS/MS检测,通过计算两份样本中内标的峰面积之比来评估样本前处理绝对回收率。本研究评估了正常、溶血、脂血和黄疸样本可能存在的基质效应,首先对这些样本进行前处理,再向前处理后的基质和纯标准溶液中加入等量的内标,并精确记录其质量,最后进行LC-MS/MS检测。通过计算基质中与纯标准溶液中内标的峰面积之比来评估相应样本的基质效应。LOD和LOQ分别定义为信噪比(signal to noise,S/N)≥3和10时能检测到的样本浓度。本研究用胎牛血清来稀释人血清样品从而获得低浓度样本,并对其检测来确定方法的LOQ和LOD。
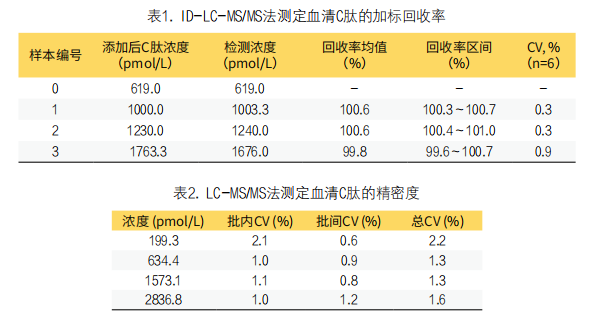
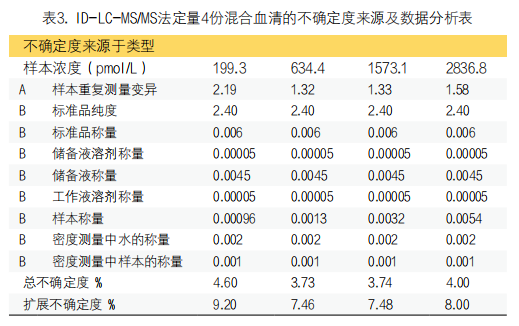
2. 加标回收率、精密度和测量不确定度:本研究通过验证方法的加标回收率来评估正确度。向同一血清样品(1.86ng/g)中加入不同量的标准溶液以制备低、中、高三种浓度水平的血清样品,对这些血清样本连续检测3天(三个批次),每次检测重复三次。对比3种加标制备血清样本的C肽浓度与原血清样本的C肽浓度,考察方法的加标回收率。本研究根据CLSI EP15-A3文件评估方法的精密度[11]。测量4份不同浓度的混合血清样本(199.3~2836.8pmol/L),每天重复检测5次连续检测五天,每个样本共得到的25个结果,采用方差分析(ANOVA)计算批内和批间以及总不精密度。同时根据国际标准化组织的测量不确定度指南,评估了该方法在四个浓度水平上的测量不确定度[12]。
七、方法对比
两种常规检测免疫方法与所建立的ID-LC-MS/MS法同时检测76份单人份血清样本(173~3612pmol/L),每个样本重复检测三次,对两者的检测结果进行普通线性回归分析和差异分析。
八、统计学分析
在本研究中,普通线性回归分析用于评估ID-LC-MS/MS方法与常规检测之间的一致性和相关性。Bland-Altman(BA)差异分析用于评估常规方法与ID-LC-MS/MS法的百分比差异。利用Microsoft Excel 2016(美国)和MedCalc统计软件(比利时)进行数据分析。
结 果
1. 样品前处理绝对回收率、基质效应、LOQ和LOD:方法的样本前处理绝对回收率在三个浓度水平(199.3、634.4和1573.1pmol/L)上分别为:78.2%~84.7%、80.6%~83.3%和79.8%~83.6%。低浓度血清样本(113.3pmol/L)在进样量为30μl时产生的平均信噪比为33.7:1(CV=5.9%,n=10);因此,方法的LOQ和LOD计算为33.3pmol/L和10.0pmol/L。正常、溶血、脂血和黄疸样品均没有明显的基质效应,这些样本的内标峰面积较纯标液的内标峰面积比平均增加0.03%。正常、溶血、脂血和黄疸血清样品较纯标液的基质效应分别为-0.45%、0.74%、-0.61%和0.45%。
2. 加标回收率、精密度和测量不确定度:方法的加标回收率见表1。三种加标血清的平均回收率和CV分别为:100.6±0.3%、100.6±0.3%和99.8±0.9%。方法在四个浓度血清水平(199.3~2836.8pmol/L)上的批内、批间和总精度分别为:1.0%~2.1%、0.6%~1.2%和1.3%~2.2%。见表2。方法在检测四个不同浓度混合血清样本时的相对测量不确定度范围为3.7%~4.6%,见表3。
3. 方法对比:两种常规免疫测定法与ID-LC-MS/MS法之间普通线性回归见图2和表4。结果显示,ID-LC-MS/MS法与免疫法有着明显的相关性,R2均大于0.98,且普通线性回归线的斜率接近1.0,但截距较大,范围为-108.7~-148.0(表4)。两种常规方法与ID-LC-MD/MS法间相对差异较小(均<10%),见图2、表4。
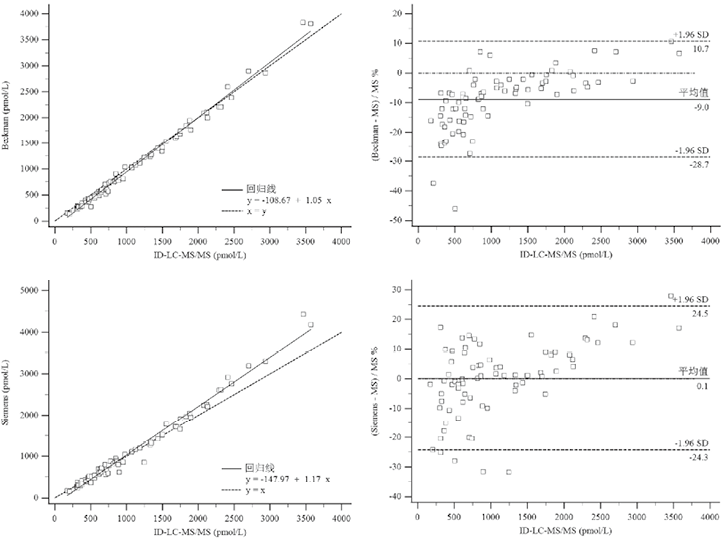
图2. ID-LC-MS/MS法与两种常规方法的普通线性回归图及差异分析图
讨 论
合适的样本前处理对ID-LC-MS/MS法的性能十分重要,其应在提取足够多的待测物质,提高检测灵敏度的同时,也应能除掉绝大部分无关物质从而减小基质效应。本研究对之前样本前处理程序进行了改进,在保证高回收率和高特异性的同时,能够简单快速地从血清中提纯C肽[13, 14]。本研究使用ZnSO4溶液代替常见的有机试剂来沉淀蛋白质,这样便无需额外的操作来降低上清液中有机溶剂的比例,从而能直接进行阴离子交换固相萃取(SPE)。同时在对其进行阴离子交换固相萃取时,利用C肽为酸性多肽的特点(等电点约为3.2)的特点,通过调节淋洗液和洗脱液的PH值,有效的提纯了C肽,并除去大量杂质。
C肽是由31个氨基酸残基组成的多肽,分子量在3020左右,不同于小分子物质,其在串联质谱中的裂解效率低下,因此需要提高其响应。本研究发现,调节串联质谱的分辨率能有效提高C肽在串联质谱中的响应,将Q1分辨率由中(Unit)调整为低(Low)可以将子离子信号增强近四倍。这可能是由于不同于小分子物质,C肽分子质量大,存在更多同位素峰的分布。当Q1分辨率为中(Unit)时,由于筛选窗口较窄,仅捕获了C肽的一小部分同位素峰,而将Q1分辨率调低后,筛选窗口变宽,允许C肽更多的同位素分布进入碰撞室,从而碰撞产生更多的子离子。
诸多研究表明,C肽具有易于吸附的特点,方法的建立过程中,通常通过添加非特异性结合蛋白(如牛血清白蛋白)来竞争性结合吸附位点,从而减少C肽的吸附[15]。本研究最初将C肽标准品配置在0.1%的牛血清白蛋白溶液中,但后续研究发现,标准品的前处理绝对回收率远低于样本的绝对回收率,这有可能是因为,在前处理过程除去了部分牛血清白蛋白,其浓度降低,不能有效的抑制C肽的吸附。因此,本研究后续使用不含人C肽的胎牛血清来制备标准溶液和内标溶液,其行为与血清样品相似,能有效的抑制C肽的吸附。
根据Westgard网站(https://www.westgard.com/biodatabase1.htm)推荐的标准,C肽常规检测方法的精密度应≤8.3%,正确度应≤7.1%。本研究所建立的ID-LC-MS/MS法精密度≤2.2%,低于常规方法不精密度标准的1/3。由于目前国际上没有相应的血清参考物质,本研究采用加标回收实验来验证方法的正确度,方法的加标回收率在99.6%~101.0%之间,其显示出来的正确度低于常规检测正确度标准的1/4。出色的回收率表明所建立方法可以完整识别并提取出样品中的待测物,并为识别的待测物提供稳定的测量信号。然而,定量的正确性还取决于校准品的溯源性。本研究中,校准品为JCTLM列表收入的C肽一级参考物质,该参考物质具有认证的浓度和不确定度并溯源到SI单位[16]。
综上所述,本研究所建立的方法精密可靠,有望用作血清C肽测定的候选参考方法。该方法在将来可用于室间质评计划和正确度验证计划材料赋值,促进中国临床实验室C肽检测的标准化。![]()
参考文献
Zimmet P, Alberti KG, Magliano DJ, et al. Diabetes mellitus statistics on prevalence and mortality: facts and fallacies, Nat Rev Endocrinol, 2016, 12 (10): 616-622.
Guariguata L, Whiting DR, Hambleton I, et al. Global estimates of diabetes prevalence for 2013 and projections for 2035, Diabetes Res Clin Pract, 2014, 103 (2): 137-149.
Leighton E, Sainsbury CA, Jones GC. A Practical Review of C-Peptide Testing in Diabetes, Diabetes Ther, 2017, 8 (3): 475-487.
Jones AG, Hattersley AT. The clinical utility of C-peptide measurement in the care of patients with diabetes, Diabet Med, 2013, 30 (7): 803-817.
Oyer PE, Cho S, Peterson JD, et al. Studies on human proinsulin. Isolation and amino acid sequence of the human pancreatic C-peptide, J Biol Chem, 1971, 246 (5): 1375-1386.
Zavaroni I, Deferrari G, Lugari R, et al. Renal metabolism of C-peptide in man, J Clin Endocrinol metab, 1987, 65 (3): 494-498.
Henriksen JH, Tronier B, Bülow JB. Kinetics of circulating endogenous insulin, C-peptide, and proinsulin in fasting nondiabetic man, metabolism, 1987, 36 (5): 463-468.
Zhou W, Deng Y, Zhao H, et al. Current Status of Serum Insulin and C-Peptide Measurement in Clinical Laboratories: Experience from 94 Laboratories in China, Ann Lab Med, 2022, 42 (4): 428-437.
Liu Z, Liu Q, Deng Y, et al. Quantitation of plasma metanephrines using isotope dilution liquid chromatography tandem mass spectrometry (ID-LC/MS/MS): a candidate reference measurement procedure and its application to evaluating routine ID-LC/MS/MS methods, Anal Bioanal Chem, 2021, 413 (30): 7509-7520.
Kinumi T, Mizuno R, Takatsu A. Quantification of serum C-peptide by isotope-dilution liquid chromatography-tandem mass spectrometry: enhanced detection using chemical modification and immunoaffinity purification, J Chromatogr B Analyt Technol Biomed Life Sci, 2014, 953-954138-142.
CLSI. EP15-A3: User verifcation of precision and estimation of bias; approved guideline - Third edition[S]. Wayne, 2014.
ISO/IEC. Guide 98–3:2008. Uncertainty of measurement - part 3: guide to the expression of uncertainty in measurement[S]. Geneva, 2008.
Stoyanov AV, Connolly S, Rohlfing CL, et al. Human C-peptide Quantitation by LC-MS Isotope-Dilution Assay in Serum or Urine Samples, J Chromatogr Sep Tech, 2013, 4 (3):
Stoyanov AV, Rohlfing CL, Connolly S, et al. Use of cation exchange chromatography for human C-peptide isotope dilution - mass spectrometric assay, J Chromatogr a, 2011, 1218 (51): 9244-9249.
Rogatsky E, Balent B, Goswami G, et al. Sensitive quantitative analysis of C-peptide in human plasma by 2-dimensional liquid chromatography-mass spectrometry isotope-dilution assay, Clin Chem, 2006, 52 (5): 872-879.
Kinumi T, Goto M, Eyama S, et al. Development of SI-traceable C-peptide certified reference material NMIJ CRM 6901-a using isotope-dilution mass spectrometry-based amino acid analyses, Anal Bioanal Chem, 2012, 404 (1): 13-21.


