


宏基因组测序生物信息学分析的临床应用与面临的问题

李波,医学博士、副主任医师、硕士研究生导师。现任解放军总医院第五医学中心检验科副主任,中国合格评定国家认可委员会(CNAS)技术评审员。现担任中国医疗保健国际交流促进会基层检验技术标准化分会常务委员兼秘书长、中华医学会病毒学分会第十届委员会青年委员、北京医学会微生物与免疫学会青年委员、北京医学会检验医学分会生化学组委员、全军仿生专业委员会青年委员、《国际检验医学杂志》审稿专家、《现代医学与健康研究&大医生》杂志编委。长期从事肝癌肿瘤标志物研究、新突发传染病病原快速诊断技术研究。承担首都临床特色应用研究专项、全军医学科技青年培育计划等多项课题,在Mol.Biol. Evol、BMC Infectious Diseases等国际期刊发表SCI论文12篇,其中以第一作者发表SCI论文6篇,中文核心期刊论文25篇,副主编专著1部,参编5部,获计算机软件著作权2项,实用新型专利1项。获得军队科技进步一等奖、医疗成果二、三等奖各一项。

杨光,医学博士、副研究员。现任职于解放军总医院第五医学中心检验科。主持全军医学科技青年培训项目一项,参与国家及军队课题3项,目前主要从事感染性疾病检测技术研究。以第一作者及共同第一作者发表SCI论文8篇。2017年和2022年先后两次赴西非执行援助塞拉利昂军事医学任务,承担临床检验及科学研究等工作。
随着测序技术的飞速发展,采用新一代测序技术(NGS)产生的数据越来越大,在实际工作中,由测序获得的数据需要进行复杂的数据解读和分析才能得到有用的信息。宏基因组NGS(metagenome next-generation sequencing,mNGS)技术被广泛应用于各种感染性疾病诊断、新发突发传染病病因分析、耐药基因检测和宿主免疫应答分析等领域[1-3]。但是与单一物种产生的NGS数据相比,mNGS产生的基因组数据要复杂得多。多数临床样本中都含有大量数量未知的物种DNA信息,且各物种的相对丰度差异很大,导致mNGS的测序数据高度分散、冗余度低。因此,针对临床微生物检测mNGS的生物信息学数据分析也很难达成一致化和标准化。2021年,为提高临床对mNGS结果的理解, 中华医学会检验医学分会发布了《宏基因组测序病原微生物检测生物信息学分析规范化管理专家共识》,根据国内外的发展现状,结合国内测序实验室常规做法,阐述生物信息学分析的规范化管理内容[4]。本文将结合共识和相关文献的内容,进一步总结生物信息学分析方法在病原微生物mNGS检测中的应用方法及临床所需要解决的问题,以期推动mNGS技术在临床微生物检测工作中更好地发展。
一、宏基因组分析的基本内容及流程
宏基因组数据的分析目前主要包括三种类别:基于序列(Reads)的宏基因组分析、基于组装(de-novo)的宏基因组分析、基于分箱(binning)的宏基因组分析。数据分析从下机原始序列开始,首先对原始序列进行去接头、质量剪切以及去除污染等优化处理。经过优化处理后的数据可以直接使用Reads与已知物种和功能数据库进行比对,从而得到每个样本的物种、功能注释信息和相对丰度。此方法最大的优势是不用进行基因组的组装,从而快速得到群落的物种与功能组成信息。但是,基于Reads与已知序列比对的方法,没办法表征大量尚未分离和测序的微生物群体。因此,将处理过的优质序列进行拼接组装和基因预测,或采用分箱将物种分开归类得到非冗余基因集,更适用于研究包含大量以前未观测到(未测序)的微生物[5, 6]。
获得可以用于分析的优质数据后,宏基因组的后续分析主要包括物种分析、功能分析、多样性分析、差异分析等。(1)物种分析:是对微生物物种进行注释和分类,就是了解每个样本中存在的微生物种类和丰度、菌群之间的差异等基本信息。常用的分析软件包括基于k-mer算法的kraken和基于标记特征分析的metaPhlAn[7, 8]。(2)功能分析:除了关注物种注释分类和丰度分析外,通常还需要分析微生物的基因功能和可能参与的通路。例如通过与KEGG、eggNOG、GO、CARD等功能注释数据库进行比较,分析菌群在生物代谢通路、分子功能、抗生素抗性基因等方面的情况[9, 10]。例如使用HUMAnN2(The HMP Unified metabolic Analysis Network 2)可以进行宏基因组学微生物代谢途径和功能分析[11]。(3)多样性分析:是指对微生物种群内或种群间的差异性和一致性进行分析,包括α多样性和β多样性分析[12, 13]。(4)差异分析:主要是采用LEFSe分析进行组间比较,通过寻找具有统计学差异的物种生物标识用于解释高维度数据,它强调统计意义和生物相关性[14]。
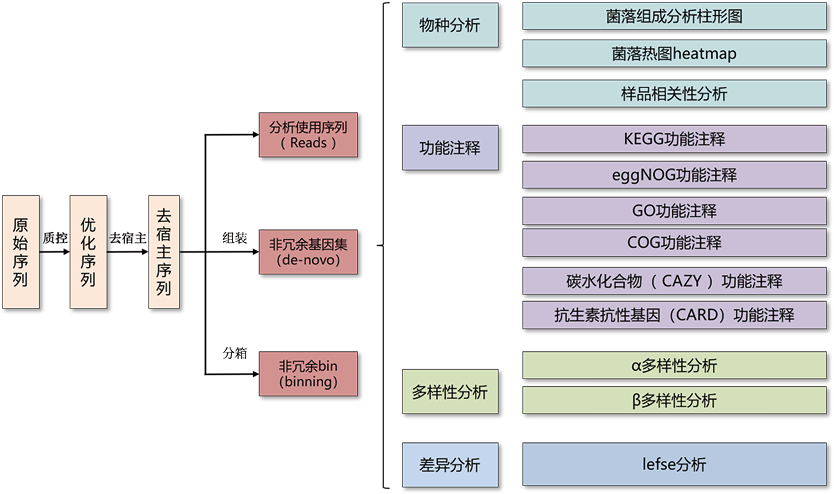
图1. 微生物宏基因组数据分析分类和基本流程
二、宏基因组数据分析能力软硬件需求
专家共识指出:生物信息分析人员应遵循 mNGS 数据分析流程,使用的分析软件应有国家药品监督管理局认证资质,从原始序列到非人源高质量序列的获得以及物种注释需要经过标签识别、测序数据质量和测序深度评估、低质量序列过滤、去人源核酸序列、高质量序列比对等环节,每一步需严格遵守上述流程及具体要求[4]。mNGS 数据分析涉及的软件如图2所示。虽然有一些NGS分析软件(如CLC Genomics Workbench)可以在Windows环境下运行,但大多数软件工具只能在Linux(Unix)环境下运行,所以服务器推荐安装Linux(Unix)操作系统,考虑到数据安全,推荐本地服务器部署,离线更新。分析软件可以通过Bioconductor平台获取,这个平台包含了超过1000个的软件包,其中有许多都是被设计用于处理NGS数据的。生信分析人员可以通过安装Anaconda或Miniconda来建立虚拟环境或下载Bioconductor提供的资源。
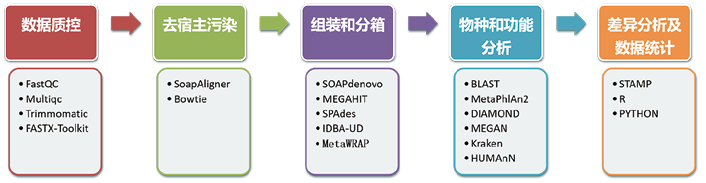
图2. 微生物宏基因组数据分析软件
在数据库选择方面,专家共识建议采用专用软件构建满足临床检测的比对数据库,其至少应包括微生物、人源及背景序列数据等。实验室应保证数据库稳定运行,及时补充新发病原体,提高数据库覆盖度,以加密形式存储于服务器中并备份,仅由授权的生物信息分析人员调用数据。对于计算能力的配置问题,主要取决于实验室需要进行分析的类型。例如大型的基因组从头组装需要高性能的集群计算系统,而对于小型项目,NGS数据分析可以选择基本的64位计算机,8GB的RAM和两个2GHz的四核处理器即可完成序列读段与参考基因组的定位运算。当然,考虑到适应未来计算需求的增加,最好配置可扩展的计算系统。实验室配置的服务器需保证单次mNGS下机数据在30min至1h内完成批量样本并行分析。同时,实验室使用的服务器只能用于mNGS,不可与其他计算需求共用。
三、宏基因组数据分析在临床微生物实验室应用的标准化
2018年美国病理协会(AMP)和美国病理学家协会(CAP)发布了《NGS生物信息流程验证标准和指南》,针对二代测序的生物信息学流程提出了17条共识,建议实验室在验证生物信息学流程时充分考虑实验室生信分析流程标准化和规范化问题[15]。共识的内容主要包括:(1)临床实验室自建NGS数据分析流程,必须进行验证,以确保实验结果的可靠性,且验证过程必须由经过培训的医学专业人员全程参与验证和监督,如果分析过程发生变更,需要进行再次验证;(2)实验室的数据分析过程必须符合行业认证和法律法规要求。例如,样本必须有唯一标识、数据分析流程中的所有数据和记录必须真实完整的保留、必须建立相关的质量控制措施确保检测质量;(3)在验证和实验过程中,可以采用计算机模拟数据,但是整个验证过程应尽可能模拟真实的实验室环境,并且模拟数据不可替代真实样本的数据验证;(4)需要充分合理地利用分析系统和软件,但是不能完全依赖软件的判断结果,需要进行人工审核和专业的数据解读。虽然这些建议主要是基于人类生殖系和体细胞突变测序分析的,但也都适用于宏基因组测序的生物信息学分析。因此对mNGS测序分析,我们同样建议对分析过程中的各个指标进行分析和评估,比如测序reads数、测序深度、reads质量等。在每次测序分析过程中,均需采用内标及阴阳性对照进行质量控制。临床实验室的mNGS分析流程应遵循ISO15189质量体系认可要求,建立符合实验室实际情况的质量管理体系并严格按照体系运行、持续改进。
四、宏基因组数据分析临床应用面临的问题及挑战
由于目前绝大部分mNGS数据分析系统和软件还是在Linux系统平台下使用,这对大部分实验室技术人员而言难度较大,如临床实验室想自己建立mNGS分析流程,需要耗费大量的资源对员工进行技能培训,并且生信分析对技术人员的素质要求也相当高,如果要完全掌握生信分析技能,涉及到数学、计算机科学、统计学等多学科。开展临床检测工作,还同时需要精通临床微生物学。所以,技术人员能力是临床mNGS开展面临的第一个挑战。其次,mNGS数据存储也是一个难题,目前的测序数据通常储存在本地服务器中或上传至云端服务器。但是涉及到患者的隐私保护问题,云端数据存储是存在一定风险的。由于mNGS数据量巨大且持续更新,如果想将数据存于本地服务器,则实验室需要考虑硬件的配置和持续升级的问题[16]。
目前的mNGS数据分析流程主要是基于科研需求开发的,和临床检测要求的标准化和准确率还有一定距离。对临床实验室而言,通常是自己制定流程方案,根据临床需求,采用特定的软件或数据库进行分析。临床数据分析同样包括原始数据的质控、去接头、去宿主基因、组装拼接、物种比对等步骤,而上述步骤比科研应用的要求更加严格,务必确保准确性和完整性,避免错误误导临床诊断。同时,临床实验室要充分利用标准的参考物质和NGS数据集来完善自己的生信分析流程。北京医院的研究团队2023年发布的支气管肺泡灌洗液(BALF)宏基因组测序数据分析自建方法评价验证方案,对实验室生信分析的流程建立具有指导意义[17]。部分公共数据库和微生物的参考基因组在持续更新,实验室需要随时追踪这些数据库的更新和发展,常用的数据库如NCBI的核酸数据库,非常全面但是错误率也相对较高。FDA-ARGOS及RVDB数据库等虽然不完整,但是可以作为通用数据库的参考和补充[18]。
这与其他临床检测项目一样,mNGS数据分析对性能验证、质量控制、样本比对等方面也有要求,这也是确保临床结果准确所必需的。所以,对临床样本的mNGS数据分析也需设置相应的阈值排除背景污染,提高真阳性率。设置的阈值可以参考以下指标:和参考数据库物种比对一致的序列数量、外部非模板质控或内部spike-in质控品、覆盖非重叠基因组区域的数量、临床样本序列丰度与阴性对照样本的对比(避免污染菌误报)。临床可以采用ROC曲线建立合适的检测阈值,但需要独立样本组进行验证[19]。
mNGS在临床实验室的应用前景广泛,尤其是对临床出现的未知病原体感染的筛查和确认,mNGS是最后可行的方案。但是,mNGS在临床的推广应用同样存在着困难,除了实验程序要求标准化和可重复外,生物信息学数据分析也至关重要。目前急需解决的问题包括人员技术能力、硬件要求、质量控制、流程标准化等。随着越来越多的临床实验室参与,mNGS正在逐渐地从科研领域转移到微生物检测领域。相信在不久的将来,mNGS也会成为精准诊断病原感染的关键驱动力和技术手段。![]()
参考文献
Han D, Li Z, Li R. mNGS in clinical microbiology laboratories: on the road to maturity. Crit Rev Microbiol. 2019, 45(5-6): 668-685.
Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation Sequencing for pathogen detection. Annu Rev Pathol. 2019, 14: 319-338.
Jing C, Chen H, Liang Y, et al. Clinical evaluation of an improved metagenomic next-generation sequencing test for the diagnosis of bloodstream infections. Clin Chem. 2021 Aug 5; 67(8): 1133-1143.
中华医学会检验医学分会. 宏基因组测序病原微生物检测生物信息学分析规范化管理专家共识. 中华检验医学杂志, 2021, 44(9): 799-807.
Nielsen HB, Almeida M, Juncker AS, et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat Biotechnol. 2014, 32(8): 822-828.
Zhang Z, Zhang L, Zhang G, et al. Deduplication improves cost-efficiency and yields of de novo assembly and binning of shotgun metagenomes in microbiome research. Microbiol Spectr. 2023, 11(2): e0428222.
Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014; 15(3): R46.
Truong DT, Franzosa EA, Tickle TL, et al. metaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods. 2015, 12(10): 902-903.
Hernández-Plaza A, Szklarczyk D, Botas J, et al. eggNOG 6.0: enabling comparative genomics across 12 535 organisms. Nucleic Acids Res. 2023,D389-D394.
Alcock BP, Huynh W, Chalil R, et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 2023, 51(D1): D690-D699.
Franzosa EA, McIver LJ, Rahnavard G, et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018,15(11):962-968.
Xiao G, Cai Z, Guo Q, et al. Insights into the unique lung microbiota profile of pulmonary tuberculosis patients using metagenomic next-generation sequencing. Microbiol Spectr. 2022, 10(1): e0190121.
Fu Y, Wu J, Wang D, et al. metagenomic profiling of ocular surface microbiome changes in demodex blepharitis patients. Front Cell Infect Microbiol. 2022, 12: 922753.
Chang F, He S, Dang C. Assisted selection of biomarkers by linear discriminant analysis effect size (LEfSe) in microbiome data. J Vis Exp. 2022, (183).
Roy S, Coldren C, Karunamurthy A, et al. Standards and guidelines for validating next-generation sequencing bioinformatics pipelines: A joint recommendation of the association for molecular pathology and the college of american pathologists. J Mol Diagn. 2018, 20(1): 4-27.
Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019, 20(6): 341-355.
Diao Z, Lai H, Han D ,et al. Validation of a metagenomic next-generation sequencing assay for lower respiratory pathogen detection. Microbiol Spectr. 2023, 11(1): e0381222..
Goodacre N, Aljanahi A, Nandakumar S, et al. A reference viral database (RVDB) To enhance bioinformatics analysis of high-throughput sequencing for novel virus detection. Msphere. 2018, 3(2): e00069-18.
Miller S, Naccache SN, Samayoa E, et al. Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 2019, 29(5): 831-842.


