


遗传性共济失调症全基因组测序与诊疗分析
一、临床病历资料
![]() 1. 一般情况:患者,王某某,女,38岁,年轻女性。
1. 一般情况:患者,王某某,女,38岁,年轻女性。
![]() 2. 主诉:头部抖动2月,慢性病程。
2. 主诉:头部抖动2月,慢性病程。
![]() 3. 现病史:患者近2个月以来出现不自主头部抖动,伴有记忆力减退、嗅觉减退,伴有动作减慢、走路不稳,觉肢体僵硬,情绪紧张时抖动加重,休息及心情放松时可以好转,但不能完全缓解,睡眠状态不抖,上述症状逐渐加重,为进一步诊治,于2023年2月2日就诊,以“头部不自主抖动待诊”收入院。患者自发病以来,精神可、食欲、睡眠可,大小便正常,体重无明显变化。
3. 现病史:患者近2个月以来出现不自主头部抖动,伴有记忆力减退、嗅觉减退,伴有动作减慢、走路不稳,觉肢体僵硬,情绪紧张时抖动加重,休息及心情放松时可以好转,但不能完全缓解,睡眠状态不抖,上述症状逐渐加重,为进一步诊治,于2023年2月2日就诊,以“头部不自主抖动待诊”收入院。患者自发病以来,精神可、食欲、睡眠可,大小便正常,体重无明显变化。
![]() 4. 既往史:10+年前发作性头痛,每次持续1天左右,有时伴有恶心、呕吐、畏光、畏声,上午轻,下午重,精神紧张时加重,休息后可好转。曾就诊,未治疗,具体不详。考虑偏头痛?
4. 既往史:10+年前发作性头痛,每次持续1天左右,有时伴有恶心、呕吐、畏光、畏声,上午轻,下午重,精神紧张时加重,休息后可好转。曾就诊,未治疗,具体不详。考虑偏头痛?
![]() 5. 其他:余无特殊,个人史、婚育史、月经史、家族史无特殊。
5. 其他:余无特殊,个人史、婚育史、月经史、家族史无特殊。
![]() 6. 查体:Bp 144/82mmHg,神志清楚,言语流利,但声音发颤,计算力、记忆力、理解力大致正常,双侧瞳孔等大,直径约3.0mm,直接、间接对光反射灵敏,双眼球各向活动可,无复视、眼震,双侧额纹、鼻唇沟对称,伸舌居中,双侧咽反射存在,四肢肌力5级,肌张力适中,四肢腱反射(++),双侧巴氏征(-),双侧额面部及肢体痛觉对称,双侧指鼻试验、跟膝胫试验稳准,头部震颤,双手偶有震颤,颈软。
6. 查体:Bp 144/82mmHg,神志清楚,言语流利,但声音发颤,计算力、记忆力、理解力大致正常,双侧瞳孔等大,直径约3.0mm,直接、间接对光反射灵敏,双眼球各向活动可,无复视、眼震,双侧额纹、鼻唇沟对称,伸舌居中,双侧咽反射存在,四肢肌力5级,肌张力适中,四肢腱反射(++),双侧巴氏征(-),双侧额面部及肢体痛觉对称,双侧指鼻试验、跟膝胫试验稳准,头部震颤,双手偶有震颤,颈软。
![]() 7. 辅助检查:(1)头颅MRI+DWI:未见明显异常。(2)脑电图:未见明显异常。(3)HOLTER:窦性心律不齐,间歇性一度房室传导阻滞。(4)心脏超声:未见明显异常。(5)血常规、肝肾功、电解质、叶酸、维生素B12、同型半胱氨酸、甲功、铜蓝蛋白、血清微量元素、OGTT、胰岛素释放未见明显异常。(6)血脂高:胆固醇6.18mmol/L,低密度脂蛋白胆固醇3.8mmol/L。(7)腰穿:颅压170mmH2O。CSF无色透明。CSF白细胞2×106/L,红细胞0,氯126.9mmol/L,糖3.58mmol/L,蛋白0.286g/L。CSF抗酸、墨汁阴性。CSF+血清:自身免疫性脑炎抗体阴性。(8)量表评估:焦虑22分,抑郁15分,MoCA18分,MMSE20分。(9)基因检测:送检未一医学全基因组测序分析,结果待出。
7. 辅助检查:(1)头颅MRI+DWI:未见明显异常。(2)脑电图:未见明显异常。(3)HOLTER:窦性心律不齐,间歇性一度房室传导阻滞。(4)心脏超声:未见明显异常。(5)血常规、肝肾功、电解质、叶酸、维生素B12、同型半胱氨酸、甲功、铜蓝蛋白、血清微量元素、OGTT、胰岛素释放未见明显异常。(6)血脂高:胆固醇6.18mmol/L,低密度脂蛋白胆固醇3.8mmol/L。(7)腰穿:颅压170mmH2O。CSF无色透明。CSF白细胞2×106/L,红细胞0,氯126.9mmol/L,糖3.58mmol/L,蛋白0.286g/L。CSF抗酸、墨汁阴性。CSF+血清:自身免疫性脑炎抗体阴性。(8)量表评估:焦虑22分,抑郁15分,MoCA18分,MMSE20分。(9)基因检测:送检未一医学全基因组测序分析,结果待出。
![]() 8. 治疗方案:(1)服用艾地苯醌、瑞舒伐他汀。(2)头部不自主抖动:阿罗洛尔片10mg Po qd,有效但不佳,10mg po bid好转。(3)焦虑状态:舍曲林片、九味镇心颗粒、右佐匹克隆治疗有效。
8. 治疗方案:(1)服用艾地苯醌、瑞舒伐他汀。(2)头部不自主抖动:阿罗洛尔片10mg Po qd,有效但不佳,10mg po bid好转。(3)焦虑状态:舍曲林片、九味镇心颗粒、右佐匹克隆治疗有效。
![]() 9. 出院诊断:特发性震颤?焦虑状态,认知障碍,高脂血症。
9. 出院诊断:特发性震颤?焦虑状态,认知障碍,高脂血症。
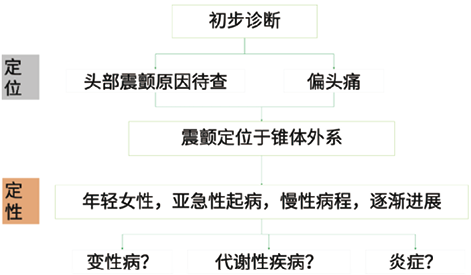
图1. 患者初步诊断情况
二、全基因测序明确诊断
![]() 1. 未一医学WGS检测结果:先证者脊髓小脑共济失调8型ATXN8基因(CAG/TAG)n三核苷酸重复数为18/70次,一个等位基因正常(正常重复数参考值≤50次),另一个异常扩展(异常重复数参考值≥50次)。
1. 未一医学WGS检测结果:先证者脊髓小脑共济失调8型ATXN8基因(CAG/TAG)n三核苷酸重复数为18/70次,一个等位基因正常(正常重复数参考值≤50次),另一个异常扩展(异常重复数参考值≥50次)。
表1. 先证者全基因组测序结果

![]() 2. 亲属验证结果:遵循先证者及家属意愿,通过PCR-毛细管电泳(FA)法对致病基因动态突变进行验证,结果显示先证者(27/109)、其父亲(27/108)、两个女儿(18/>109、23/176)存在异常扩展,其母亲、两个弟弟及弟弟子女未见异常。
2. 亲属验证结果:遵循先证者及家属意愿,通过PCR-毛细管电泳(FA)法对致病基因动态突变进行验证,结果显示先证者(27/109)、其父亲(27/108)、两个女儿(18/>109、23/176)存在异常扩展,其母亲、两个弟弟及弟弟子女未见异常。
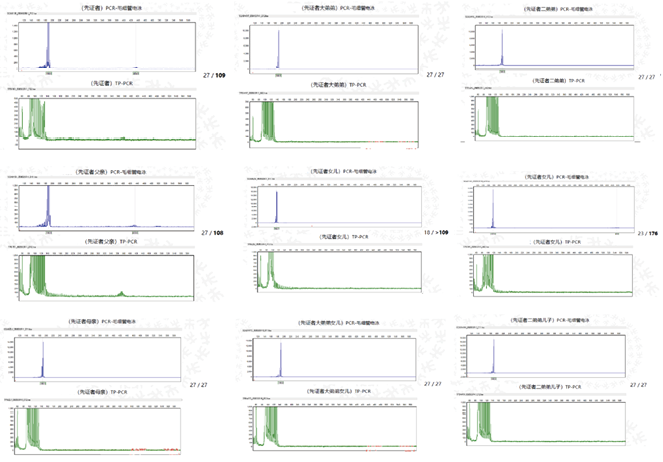
图2. 先证者家系验证结果
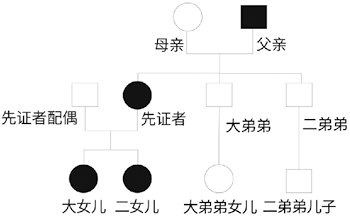
图3. 先证者家系谱图
![]() 3. 最终诊断:遗传性共济失调—脊髓小脑共济失调8型(SCA8)。
3. 最终诊断:遗传性共济失调—脊髓小脑共济失调8型(SCA8)。
三、疾病概述
遗传性共济失调(Hereditary Ataxia,HA)是一大类具有高度临床和遗传异质性、病死率和病残率较高的遗传性神经系统退行性疾病,约占神经系统遗传性疾病的10%-15%。脊髓小脑共济失调(Spinocerebellar Ataxia SCA)是遗传性共济失调的主要类型,是一组由基因突变导致小脑、脑干、脊髓退行性变,以进行性运动协调功能减退、平衡失调为主要临床表现的神经系统遗传性疾病,患者多于中青年发病,其病理改变主要是脊髓、小脑和脑干的神经元变性脱失与胶质增生,临床特征为小脑性共济失调、构音障碍、眼球运动障碍、锥体束征、锥体外系表现、肌萎缩和痴呆等。
![]() 1. 临床表现:(见表2)
1. 临床表现:(见表2)
表2. 遗传性共济失调临床表现

![]() 2. 遗传模式与常见型别:SCA最初定义和经典类型SCA为常染色体显性遗传,因相应基因外显子(CAG)三核苷酸拷贝数异常重复扩增产生多谷氨酰胺(PolyQ)所致。后来也发现了常染色体隐性遗传、X连锁遗传和线粒体遗传(NARP、MERRF以及CoQ10缺乏)的类型,见表3、表4。目前已发现数十种SCA致病基因如表1,但还有部分类型未找到明确致病基因。
2. 遗传模式与常见型别:SCA最初定义和经典类型SCA为常染色体显性遗传,因相应基因外显子(CAG)三核苷酸拷贝数异常重复扩增产生多谷氨酰胺(PolyQ)所致。后来也发现了常染色体隐性遗传、X连锁遗传和线粒体遗传(NARP、MERRF以及CoQ10缺乏)的类型,见表3、表4。目前已发现数十种SCA致病基因如表1,但还有部分类型未找到明确致病基因。
表3. 遗传性共济失调遗传模式

表4. 常见SCA型别对应基因、致病性扩增、蛋白、遗传模式、发病年龄、临床表现及MRI情况
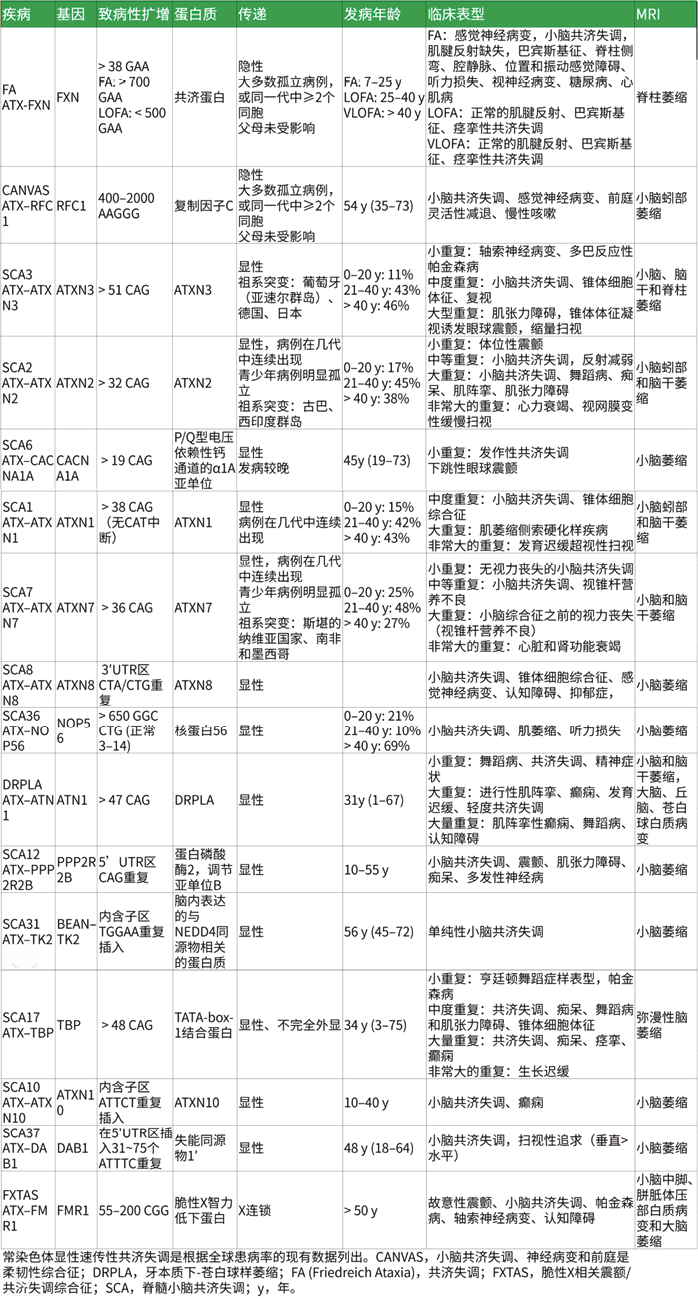
其中,SCA8临床表现包括步态和肢体共济失调、扫描构音障碍,其特征是言语缓慢、眼球运动异常,同是也可能存在上运动神经元表现(腱反射亢进、痉挛状态和Babinski征),锥体外系体征(震颤、肌张力障碍和伴或不伴帕金森样特征),脑干体征(吞咽困难和咳嗽反射差),感觉神经病变(肢体远端感觉丧失和腱反射丧失),认知功能障碍等。但对于CTA/CTG(≥71-1300)变异类型的外显率<100%,即并不是所有遗传了该扩增突变的人都会继续发展为该疾病,尽管家族中其他人可能也携带该基因变异,但患者可能是唯一出现该疾病症状的成员。
![]() 3. 基因检测策略:高度疑似SCA患者可优先进行相关基因动态突变的检测,对于FA法检测阴性的疑似遗传性共济失调患者,可进一步进行Panel或WES检测,对非动态突变引起的其他类型共济失调进行排查,如发作性共济失调(EA)和遗传性痉挛性共济失调(HSA)等。对于线粒体相关,需排查线粒体环基因变异。对于方向不明,条件允许者可行WGS检测,可一次性检测提示SNV、InDel、CNV、SV、内含子变异、动态突变、UPD等多种变异类型。
3. 基因检测策略:高度疑似SCA患者可优先进行相关基因动态突变的检测,对于FA法检测阴性的疑似遗传性共济失调患者,可进一步进行Panel或WES检测,对非动态突变引起的其他类型共济失调进行排查,如发作性共济失调(EA)和遗传性痉挛性共济失调(HSA)等。对于线粒体相关,需排查线粒体环基因变异。对于方向不明,条件允许者可行WGS检测,可一次性检测提示SNV、InDel、CNV、SV、内含子变异、动态突变、UPD等多种变异类型。
![]() 临床高度怀疑SCA:FA+NGS。
临床高度怀疑SCA:FA+NGS。
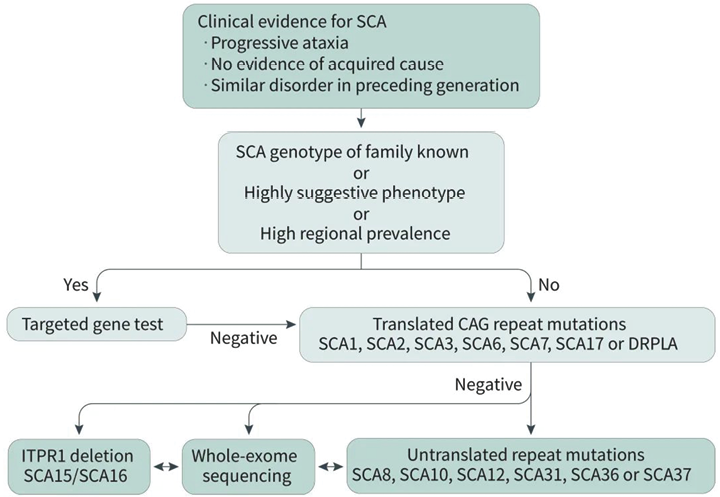
图4. 核基因相关变异推荐策略
![]() 无明确方向和/或怀疑线粒体相关共济失调:WGS、mtDNA全长测序+WES/Panel。
无明确方向和/或怀疑线粒体相关共济失调:WGS、mtDNA全长测序+WES/Panel。
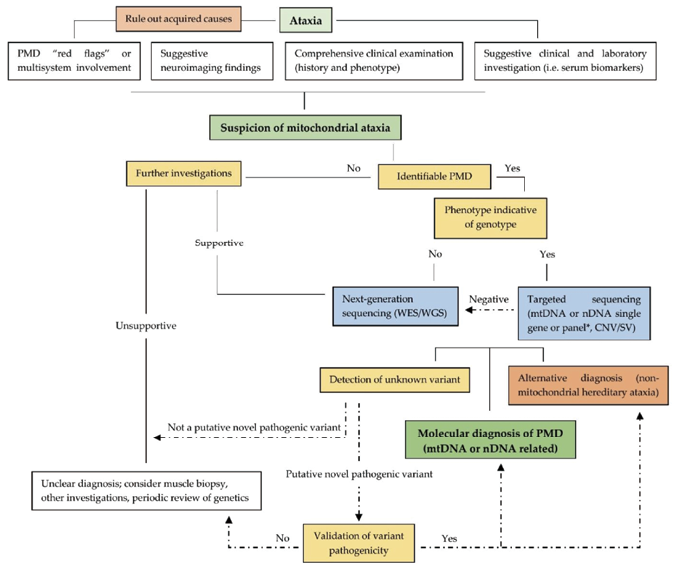
图5. 线粒体相关基因变异推荐策略
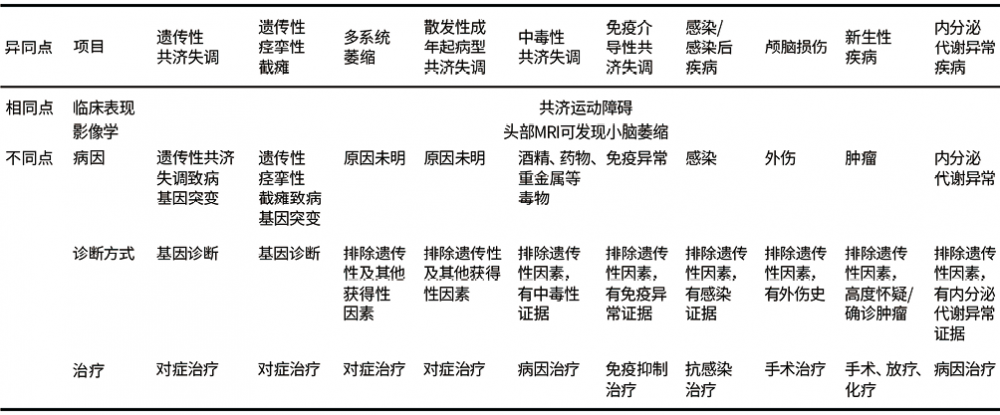
![]() 4. 鉴别诊断:应与其他遗传性及非遗传性因素所致的共济失调鉴别。(1)其他遗传性因素所致的共济失调:需通过基因诊断与遗传性痉挛性截瘫(HSP)复杂型鉴别。(2)非遗传性共济失调:包括非遗传性神经退行性共济失调及其他获得性共济失调。
4. 鉴别诊断:应与其他遗传性及非遗传性因素所致的共济失调鉴别。(1)其他遗传性因素所致的共济失调:需通过基因诊断与遗传性痉挛性截瘫(HSP)复杂型鉴别。(2)非遗传性共济失调:包括非遗传性神经退行性共济失调及其他获得性共济失调。
前者主要包括多系统萎缩(MSA)、散发性成年起病型共济失调(SAOA),其中MPA-C型以往称为橄榄体-桥脑-小脑萎缩(OPCA),是鉴别的重点。
后者主要包括中毒性共济失调(酒精、药物、重金属等所致)、免疫介导性共济失调(多发性硬化、副肿瘤综合征等)、感染/感染后疾病(小脑脓肿、小脑炎等)、颅脑创伤、新生性疾病(小脑肿瘤、转移性肿瘤等)、内分泌代谢异常(甲状腺功能减退等)等(表5)。
表5. 共济失调鉴别诊断方案
![]() 5. 治疗:目前尚无能够完全阻止病情进展的方案,尚无有效的病因治疗,临床上仍以对症和支持治疗为主,许多药物治疗尚缺乏循证医学的证据,以临床经验治疗为主,主要目标是减轻症状、延缓病情进展,改善日常生活自理能力(表6)。
5. 治疗:目前尚无能够完全阻止病情进展的方案,尚无有效的病因治疗,临床上仍以对症和支持治疗为主,许多药物治疗尚缺乏循证医学的证据,以临床经验治疗为主,主要目标是减轻症状、延缓病情进展,改善日常生活自理能力(表6)。
表6. 共济失调治疗方案

四、遗传咨询
![]() 1. 家系中患者做基因检测明确致病基因,有时需要家系成员做对照。
1. 家系中患者做基因检测明确致病基因,有时需要家系成员做对照。
![]() 2. 具有遗传风险的家系成员在怀孕之前,检测是否携带致病突变,该类疾病的预防重点在于遗传咨询,产前诊断或胚胎植入前诊断是目前有效控制发病的最佳手段。
2. 具有遗传风险的家系成员在怀孕之前,检测是否携带致病突变,该类疾病的预防重点在于遗传咨询,产前诊断或胚胎植入前诊断是目前有效控制发病的最佳手段。
![]() 3. 常染色体显性遗传家系后代,未发病+未成年,不建议做基因检测。
3. 常染色体显性遗传家系后代,未发病+未成年,不建议做基因检测。
![]() 4. 部分SCA患者没有明确家族史。
4. 部分SCA患者没有明确家族史。
(1)父母发病以前因故死亡;
(2)父母为轻症患者或中间型患者,不自觉有病(经细致查体及基因检测可明确);
(3)亲代与子代表型差异大,不被认为是同一疾病;
(4)患者为新发突变。![]()
致谢:本案例由山西白求恩医院神经内科 张瑜 提供
参考文献
Thomas K, Caterina M, Henry LP. Spinocerebellar ataxia.Nat Rev Dis Primers. 2019 Apr 11; 5(1): 24.
Mitochondrial Ataxias: Molecular Classification and Clinical Heterogeneity. Neurol Int. 2022 Apr 2; 14(2): 337-356.
中华医学会神经病学分会神经遗传学组. 遗传性共济失调诊断与治疗专家共识[J]. 中华神经科杂志, 2015, 48(6): 459-463.
https://www.ncbi.nlm.nih.gov/books/NBK1138/


