


应加强血栓与止血领域基础理论与临床研究

张真路,医学博士,主任技师,教授。现任武汉亚洲心脏病医院检验医学中心主任;武汉亚心总医院检验医学中心主任;新疆心脑血管病医院检验科主任。主要学术兼职:中国心胸血管麻醉学会临床与检验分会副主任委员,中国医师协会检验分会心脑血管疾病专业委员会副主任委员,中华医学会检验学会武汉市检验分会副主任委员,武汉非公医疗机构协会检验专业委员会主任委员,《中华心血管病杂志》(网络版)编委,IFCC心脏标志物委员会(C-CB)通讯委员。
近几年血栓与止血领域发展很快,新进展也较多。尽管ISTH已经有50年的历史,但从2019年后,其会议由每两年一次,改为每一年举办一次。这充分说明该领域的发展速度及关注程度的增加。特别是COVID-19疫情在全球肆虐,使医学界更加关注炎症或感染因素通过机体免疫系统与出凝血相关因素相互“纠缠”引起机体免疫和出凝血系统紊乱,导致机体重要器官功能损伤直至死亡。新冠病毒引发机体“细胞因子风暴”令人印象深刻。新冠可能是“一时”的,但病原微生物引起机体免疫功能紊乱导致的机体一系列的损伤机制会永远存在。由“炎症性血栓症(thromboinflammation)”或“免疫性血栓症(Immunothrombosis)”引发的多器官衰竭综合征(Multiple organ dysfunction syndrome,MODS)、shock、SIC/DIC直至死亡这条病理生理机制形成“通道”已十分明确,关键是我们要提前发现/预判,阻止病程进展或死亡。在这方面我们有许多工作要做,关键是不能把炎症/感染与出/凝血割裂考虑。
血栓与止血检验是帮助临床尽快摆脱“危急局面”的“刚需和利器”,是检验专业非常迫切和重要的“发力点”。本文就笔者所关注的血栓与止血领域新进展和研究热点加以论述,供大家参考。
一、关于细胞基础凝血理论(Cell-based coagulation model)
传统理论认为,凝血级联反应是血浆凝血蛋白通过两种不同的途径(即内源性途径和外源性途径)顺次激活,最终汇入共同途径。该凝血蛋白级联模型解释了不同的血浆凝血因子是如何被激活并相互作用的。其与凝血常规项目如aPTT和PT/INR测定有很好的相关性。INR和aPTT在评估华法林(II、VII、IX、X因子的竞争拮抗剂)和普通肝素(通过抗凝血酶III使凝血酶和Xa因子失活)的抗凝作用监测方面非常有用。但随后也发现传统凝血蛋白级联模式和常规凝血测试存在一些无法解释的现象。如在缺乏全身性抗凝血酶的情况下,INR和aPTT升高可能不是许多患者出血或血栓形成的可靠预测指标,包括创伤、败血症或肝病患者。而在危重出血相关的临床观察和研究发现,经验性新鲜冷冻血浆:血小板:红细胞的1:1:1输注策略的效果非常好;在危重出血患者中使用粘弹性和血小板功能测试(如TEG)用于治疗危重出血控制效果不错。另外,临床观察不支持传统血浆凝血蛋白级联反应所概述的内在和外在途径的独立性[1, 2]。例如,尽管aPTT显著延长增加了人类或小鼠出血的风险,但内源性凝血途径早期部分某些凝血因子(如因子XII、高分子量激肽原或前激肽释放酶)的缺乏与出血风险增加无关。aPTT和INR测试最多只能反映出体外某些(促)凝血因子的活性,而不是全部。无论是内源性途径还是外源性途径本身都不足以实现止血,单独缺乏因子VIII(血友病A)、因子IX(血友病B)或因子VII,每一种都能引起严重出血[3]。口服直接抗Ⅹa(如阿哌沙班和利伐沙班)和抗凝血酶(如达比加群)药物抑制单一凝血因子也足以实现全身抗凝,但对INR和aPTT具有不可预测和可变化的影响作用。因此,人们认识到传统的血浆凝血蛋白级联模型存在不足,需要新的理论来解释上述现象。
20多年前有人提出“细胞基础凝血模型”。他们发现离心血液样本血浆中含有所有正常的凝血因子,但没有血细胞(血小板和红细胞),如果没有激活剂(activating agents),就不能形成固体凝块。Hoffman等人在研究重组活化因子VIIa在止血中的作用机制时发现,活化因子VIIa可激活活化血小板上足够的因子X,从而恢复血小板表面凝血酶的生成[4]。Hoffman等人假设,VIIa因子活性在血小板表面的定位意味着该药物只能作用于组织损伤部位,这就解释了其安全性和有效性,以及即使在血小板减少和血小板功能缺陷患者中也能起到止血作用。他们随后提出了一个模型[5],其中凝血是由细胞表面的特性调节的,并且凝血不是作为级联发生的,而是存在三个重叠发生阶段。凝血首先发生在携带TF的细胞上,当少量凝血因子和有限数量的血小板被激活时发生扩增,增殖发生于活化血小板改变后的磷脂膜表面发生凝血酶爆发的最后阶段[6]。
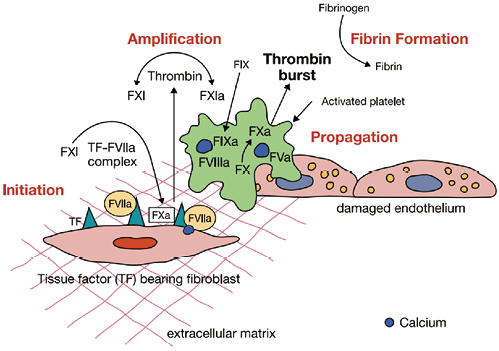
图1. 基于细胞基础的凝血模型
携带组织因子(TF)的细胞和血小板的重要性与它们如何激活凝血酶有关。在初始阶段,涉及到携带组织因子(TF)的细胞。TF-factor VII复合物产生少量因子IXa、Xa和凝血酶。在放大阶段,涉及血小板。活化血小板(包括活化GP IIb/IIIa受体),内皮细胞释放vWF及血小板表面V,VIII,XI激活。在增殖阶段,则涉及活化的血小板。在血小板表面产生活化因子Xa、IXa-FVIIIa复合物和凝血酶原(因子Xa-Va)复合物,导致凝血酶大量生成,纤维蛋白原通过活化的GP IIb/IIIa受体连接活化的血小板。
在基于细胞的凝血模型中,携带TF的细胞和血小板是两个关键的细胞相关(或细胞)成分,凝血酶和纤维蛋白原是与这些细胞相关成分协同实现止血的主要凝血蛋白[6, 7]。携带TF的细胞(如血管平滑肌细胞、外层成纤维细胞和周细胞)通常不与血浆凝血因子接触。与因子VII一起,受损血管壁上携带TF的细胞在损伤部位激活因子X和因子IX,触发凝血的起始阶段。Xa因子产生少量凝血酶(在扩增阶段),其通过血小板蛋白酶激活受体(特别是PAR-4)强烈激活血小板,导致促凝磷脂(磷脂酰丝氨酸和磷脂酰乙醇胺)在血小板膜上表达,其形状改变(从盘状变为不规则球体,具有长假足,以增加其表面积,方便血小板间连接);在凝血扩展阶段由凝血酶原产生大量凝血酶。凝血酶随后激活更多的血小板和因子XIII(一种纤维蛋白稳定因子),它连接和稳定纤维蛋白聚合物,并保留红细胞以提供血液凝块的强度和稳定性(形成红细胞、纤维蛋白和血小板塞的复杂结构)。
这种基于细胞的凝血模型非常强调含TF细胞和血小板,以及它们如何通过作为催化剂激活不同的凝血因子(导致反应速率增加千倍)而相互作用,凝血酶提供反馈回路来激活更多的血小板,然后由纤维蛋白原和血管性血液病因子聚集。此外,最近的其他研究表明,第三种细胞成分即红细胞可能也很重要,它与血小板和纤维蛋白形成一种不可渗透的复杂多面体结构,从而在不阻塞血管的情况下实现止血。这也许可以解释为什么维持一定的红细胞压积阈值可能在最严重的危及生命的重症出血中使机体存活下来[8]。
基于细胞的凝血模型代表了最具凝聚力的科学框架,在此框架上我们可以理解和管理危重出血期间的凝血问题。并解释了在当前凝血管理策略中应用基于细胞模型的潜在优势。基于细胞的凝血模型建立在传统凝血模型的基础上,解释了许多传统模型无法解释的问题。从实际应用的角度来看,应用基于细胞的凝血模型将支持(i)在相关检测结果未报告前,针对严重的危重出血使用经验性1:1:1输血策略,(ii)使用粘弹性试验指导纤维蛋白原和血小板的输注。(iii)在使用抗血小板药物治疗或怀疑血小板功能障碍的患者中,使用选定的血小板功能试验排除血小板功能障碍并指导血小板输注[9]。
需强调的是,无论我们采用基于细胞的凝血模型还是传统的凝血级联框架,全血细胞计数和凝血试验,特别是血小板计数和血浆纤维蛋白原浓度,都会保持高度的临床相关性,并应成为危及生命的危重出血患者综合管理方案的一部分。组蛋白参与了基于细胞的凝血模型的3个阶段:起始、扩增和增殖[9](图3)。
二、关于白细胞捕获网(NETs)在炎症与出凝血管控中的临床价值
1996年,Takei[10]等人发现了一种不同于细胞凋亡和坏死的细胞死亡途径。他们发现中性粒细胞多分叶状核融合,染色质紧凑结构减少,然后核膜破裂,但胞质内细胞器保持完好,3小时后,细胞外膜被破坏。2004年,布林克曼等人[11]首次报道了中性粒细胞喷射核染色质和杀菌蛋白对微生物反应的壮观图像。这种外化的染色质可以缠绕细菌,以防止其蔓延,这些外排结构被称为中性粒细胞外陷阱/捕获网(neutrophil extracellular traps,NETs)。NETs释放使捕获细菌的能力增加了三到四倍,且独立于巨噬细胞。NETs释放的这种独特的细胞死亡形式,整体称为“网状沉着症(NETosis)”,即NETs形成的生物学过程,为NETosis。
NETs是由于染色质解缩和扩散而释放的DNA结构,因此其体积是浓缩染色质体积的三到五倍。NETs中线粒体DNA的丰度是核DNA的100,000倍。有几种蛋白质附着在NETs上,包括组蛋白和初级和次级颗粒的30多种成分,其中具有杀菌活性的成分包括弹性酶、髓过氧化物酶、组织蛋白酶G、乳铁蛋白、五角蛋白3(pentraxin 3)、明胶酶、蛋白酶3、LL37、肽聚糖结合蛋白,以及其他具有杀菌活性的成分及破坏毒力因子。NETosis由DNA、组蛋白和丝氨酸蛋白酶组成会加剧血管炎症和凝血。抑制NETosis将有利于预防NETs在炎症中的毒副作用,减少病理性血栓的发生。可能最常用的NETs抑制剂是DNase,尽管它不会阻止NETs的生产,但会破坏NETs的结构。
注:a|显示NET形成步骤的荧光显微镜照片。b|相关事件对应的示意图。活化会导致活性氧簇的形成(步骤1)。然后核膜开始解体成一连串的囊泡,颗粒的完整性逐渐丧失(步骤2)。然后,细胞核失去小叶,核物质填满细胞的大部分区域,与颗粒的内容物混合(步骤3)。在最后阶段,核和颗粒的完整性完全丧失。其表现于a中步骤3的荧光通道的均匀重叠。细胞核向外聚集、收缩并最终释放NET(步骤4)。c|步骤2中性粒细胞的透射电子显微镜照片。箭头表示核膜解体,使核质与细胞质融合。其也在步骤2中的部分均一染色图案中显示。
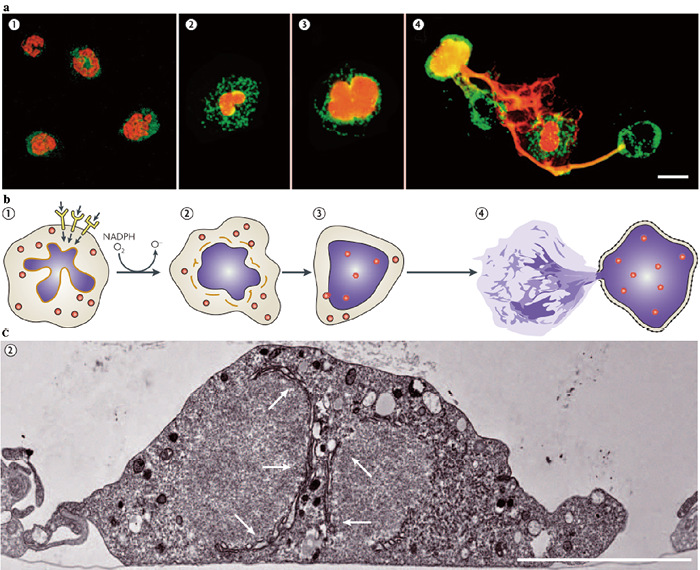
图2. NETs形成的步骤
到目前为止,已知有的三种模型NETosis。(A)自杀性网织病(suicidal NETosis),持续2-4小时且是描述最清楚的模型。其为一种细胞自杀性疾病状态,需要膜破裂和失去中性粒细胞常规功能,如白细胞募集、趋化和吞噬功能。(B)活力性网织症(In vital NETosis)。该病变过程中性粒细胞在5-60min内释放网状DNA而不表现出核或质膜的丢失,它不依赖于ROS和Raf/Merk/ERK途径。(C)最后一种类型是(vital NETosis),线粒体DNA的释放依赖于ROS,并在GM-CSF和脂多糖刺激下产生[11]。微生物及其产物、免疫复合物、自身抗体、细胞因子(IL-8、肿瘤坏死因子、I型和II型干扰素)以及其他刺激因素可引起NETosis。NETs可能是一把双刃剑,其作为有效的抗菌防御手段,但也可能是具有免疫效应和促炎作用的分子来源。在易感个体中,这些分子可能会促进组织损伤和自身免疫。虽然NETs捕获病原体,可能对感染起到有益的作用。但越来越多的证据表明,NETs在许多炎症性和血栓性疾病中是有害的,包括败血症、冠状动脉疾病、以及微血管和深静脉血栓形成。NETs可能在非感染性疾病中发挥作用,包括但不限于系统性红斑狼疮(SLE)、类风湿性关节炎(RA)、糖尿病、动脉粥样硬化、血管炎、血栓形成、癌症、伤口愈合和创伤。总之,NETs可损害宿主组织,促进自身免疫的发展,并导致其他功能障碍,包括转移、血栓形成和不适当的凝血发生(图3)[12]。
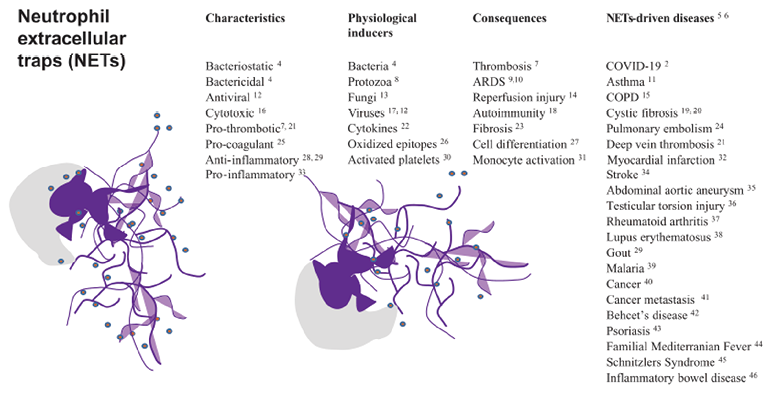
图3. 激活的中性粒细胞形成NETs(紫色),以及NETs驱动(NETs-driven diseases)的相关疾病。
在过去十多年里的研究表明,NETs有害作用通常与夸大的免疫反应有关。单个器官的过度炎症可导致远端器官损伤,从而进一步使炎症分子的释放永久化。损伤相关分子模式(DAMP)能够诱导NETs形成,并作为NETs组分产生一个致命性炎症循环,导致器官损伤并随后发生远处器官损伤。此时NETs将天平倾斜到炎症一侧,导致压倒性的免疫反应,这些过度的反应可能会影响到远处的器官并导致全身炎症,或细胞因子风暴,最终导致多器官功能障碍甚至死亡。
急性心肌梗死期间,凝血酶激活的血小板与斑块破裂部位的中性粒细胞相互作用,导致局部NETosis和组织因子的激活。组织因子是一种在内皮细胞和白细胞中表达的启动凝血和凝血酶形成的蛋白质。实验证据表明,NETs参与了自身免疫性和炎症性疾病的发病机制,并被认为参与了肾小球肾炎、慢性肺部疾病、败血症和血管疾病。一些修饰NETosis的分子(如MPO、dsDNA、组蛋白)是系统性自身免疫性疾病的自身抗原,如抗中性粒细胞胞浆抗体(ANCA)阳性的小血管炎和系统性红斑狼疮(SLE)。越来越多的证据表明NETosis在癌症中起到了作用[14]。
帕拉塞尔苏斯写道:“万物都有毒,所谓非毒,只有剂量允许某些东西不毒”。机体通过激活先天免疫系统对各种伤害或感染做出反应。它的任务是切换到炎症状态以对抗入侵者,限制或修复损伤,最终化解炎症状态并切换回稳定状态。NETs既可以抗击疾病,也可以致病,具体取决于地点、时间和剂量。在正确的时间和正确的地点处理太多或不处理NETs是致病的。NETs凝聚的形成量决定“好”或“坏”结果。如NETS参与及时的血凝块形成,但如果存在过多,它们会引发大量凝血问题,从而阻止器官的血液供应,导致严重的缺血。静脉血栓主要发生在血流量减少几个小时内,NETs紧密附着在内皮细胞上作为刺激血栓形成的支架。
确定NET形成对血栓性疾病进展的影响以及作为疾病活动性的生物标记物将有临床指导意义。NETs研究的一个相关和新兴领域是NETs和血栓形成之间的关系。NETs在血栓形成中的确切作用是什么?它们是否像纤维蛋白一样有助于血栓的稳定?它们是否与血栓形成后综合征有关?血管内NET(intravascular NETosis)释放优化了对血液中细菌和病毒的捕获。血管内NETosis也可能导致免疫血栓形成。NETs可能在血管壁损伤和新的细胞类型在血栓中的募集中发挥作用,包括用于血栓血管形成的内皮细胞。带有相关NET结构的血栓更坚硬,渗透性更差。在血栓形成过程中,NETs片段会出现在循环中,这可能是活动性血栓性疾病的有用生物标志物,应仔细研究,因为它们可能揭示更多关于NET生成和降解过程的信息。
组织损伤、炎症和感染会增加静脉血栓形成的风险,这一概念已经确立,即免疫性血栓形成。过度的炎症反应与干扰素、白介素类、肿瘤坏死因子、趋化因子水平升高有关,被称为细胞因子风暴,导致全身性炎症进展,血栓形成的风险增加。
到目前为止,NETs相关的抗炎功能或有助于消退炎症的因素还知之甚少。然而,识别代表这个圈子中关键角色的那些成分,也可能提供潜在的诊断与治疗目标,要中断这一促炎症循环。NETs/NETosis值得我们深入研究和探索,特别是如何快速与方便定量检测!
三、关于thromboinflammation和Immunothrombosis的关系
由于COVID-19的全球大流行,使我们更加关注“细胞因子风暴”这一病理生理学概念,并由此引发对thromboinflammation(炎症性血栓症)和Immunothrombosis(免疫性血栓症)这两个病理性专有名词的极大关注。其实,在过去二十年,为了描述由免疫和炎症反应引起的血栓形成和凝血障碍反应,文献交替使用炎症性血栓和免疫性血栓来描述炎症和血栓形成之间的联系。
thromboinflammation一词来源于炎症和血栓形成,是用来描述病理生理的扰动导致血管内皮损伤和/或抗血栓和抗炎功能的损失(细胞和体液炎症机制都被激活)。在急性感染中,炎症性血栓可能最终导致微血管血栓形成,这是该疾病的标志,如COVID-19,缺血再灌注损伤、创伤、器官移植排斥反应和体外循环后,并可发展为DIC。
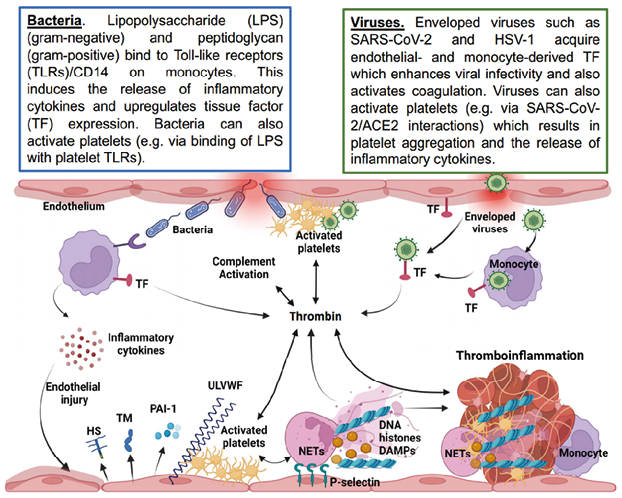
图4. 感染诱发炎症性血栓的常见机制
细菌和病毒通过不同的机制诱导组织因子的表达(如上方框所述)。组织因子启动小板和补体级联。炎性细胞因子如肿瘤坏死因子α、白细胞介素-6、白细胞介素-1和前列腺素促进内皮细胞损伤。损伤的内皮细胞表现出血栓前状态,脱落天然抗凝剂如硫酸肝素和凝血调节素,释放抑制纤维蛋白溶解的纤溶酶原激活物抑制剂-1,并从韦贝尔-帕拉德小体释放超大型vWF和p-选择素。超大型vWFF和p-选择素分别介导血小板和中性粒细胞粘附。活化的血小板诱导NETs的释放,提供一个支架来捕获血小板和红细胞。NETs成分,如胞外DNA,组蛋白和损伤相关的分子模式触发凝血,激活血小板和损害纤维蛋白溶解。严重细菌和病毒感染的最终结果是以炎症性血栓为特征的血管闭塞。
Immunothrombosis,指先天(宿主)免疫的内在效应途径,激活全身炎症和止血,通过凝血酶和纤维蛋白的产生来定位和固定感染源。作为减轻微生物侵袭炎症反应的一部分,微循环血栓形成也会造成多器官损伤。其是机体宿主防御机制,以促进微生物遏制和消除微血管血栓反应。与炎症性血栓类似,这种反应需要体液和细胞激活,通过因子XII和补体激活的接触激活途径,以及随后产生的额外促炎介质,包括NETs。因此,接触途径激活可能代表了先天免疫的生理机制,并且在SIC和急性感染中也至关重要。
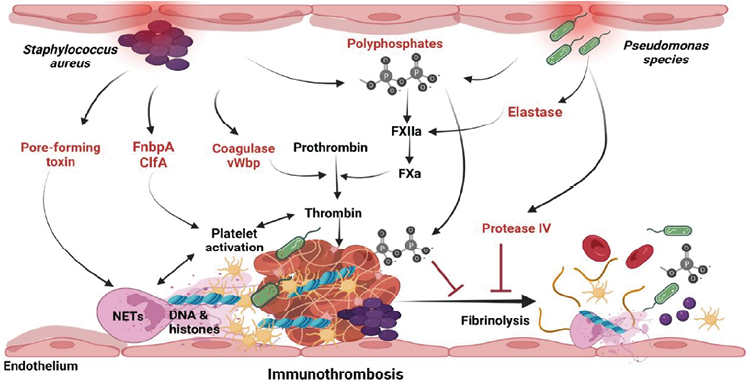
图5. 金黄色葡萄球菌和假单胞菌感染对止血的调节作用
严重的细菌感染可以通过多种机制调节止血。细菌产生的长链多磷酸盐(≥500个磷酸单位)促进FXIIa介导的凝血激活。多磷酸盐也能与纤维蛋白凝块结合,并使凝块抵抗纤维蛋白溶解。金黄色葡萄球菌分泌凝血酶和血管性血友病结合蛋白,其以非酶方式激活凝血酶原至凝血酶。金黄色葡萄球菌也分泌纤维连接蛋白A和凝块因子A,它们能激活血小板。关于NETosis,金黄色葡萄球菌分泌成孔毒素(如Panton-Valentine leucocidin),其诱导独特而快速(5-60分钟)的NETosis形式,以不依赖氧化剂的机制发生。弹性蛋白酶是假单胞菌的主要毒力因子,在凝血级联中激活FXII。假单胞菌还分泌蛋白酶IV,通过降解纤溶酶原和纤维蛋白原来抑制纤维蛋白溶解。金黄色葡萄球菌和假单胞菌严重感染的结果是免疫性血栓形成,其失调可能导致血管闭塞和炎症性血栓。
炎症性血栓和免疫性血栓有许多相似之处,但不应互换使用,即使它们在过去被用作同义词。尽管免疫血栓形成本身是一种保护性的、抗菌的机制,旨在局部抑制包括细菌和真菌在内的病原体,但正如在炎症性血栓中观察到的那样,其失调和全身恶化最终可能对宿主有害[15]。不管是thromboinflammation还是Immunothrombosis,检验专业要更加坚信“炎症与感染和出血与止血”是一对相互“诅咒为虐”的魔鬼,它们彼此相互影响,相互促进。
四、关注创伤诱导的凝血功能障碍和败血症诱导的凝血功能障碍及弥散性血管内凝血的不同病理生理学形成机制与彼此相互关系
止血是对各种损伤的基本反应,凝血对宿主防御系统至关重要,但反应过度或失调将会对机体造成严重伤害。针对不同的损伤机制,我们需关注创伤诱导的凝血功能障碍(trauma-induced coagulopathy,TIC)、败血症诱导的凝血功能障碍(sepsis-induced coagulopathy,SIC)和弥散性血管内凝血(disseminated intravascular coagulation,DIC)的不同病理生理学形成机制[16]。
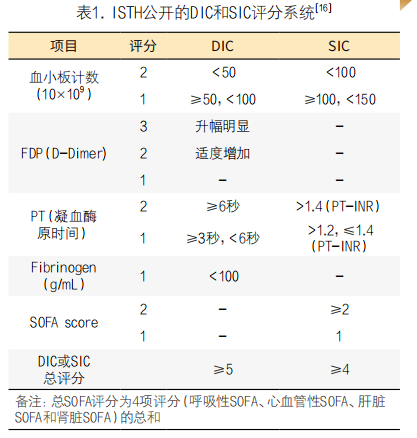
2016年,脓毒症被重新定义为“宿主对感染反应失调导致的危及生命的器官功能障碍”。ISTH DIC科学与标准化委员会将SIC定义为“感染诱导的器官功能障碍和凝血功能障碍”,并公布了由血小板计数、PT-INR和序贯器官功能衰竭评估评分组成的诊断标准[17]。TIC定义为“创伤性凝血激活,从止血受损发展到过度血栓形成,伴有失衡的纤维蛋白溶解”。尽管TIC的诊断标准尚未确定,但基于症状的临床诊断和紧急治疗手段应先于基于实验室检查的诊断。
DIC可因不同原因发生,但通常发生在败血症之后。大约25%的严重受伤患者到达医院时已发生TIC,这些患者最初表现为止血功能受损和出血表型,随后可进展到血栓形成前阶段。外伤性损伤后,大量失血、组织损伤和高纤溶导致无效止血。这种最初的止血受损持续到手术或其他治疗策略,不仅可以阻止出血的原因判断,而且还可以进展到血栓形成前和低纤溶状态,也称为纤溶性关闭[16]。
DIC可因脓毒症、创伤、血液恶性肿瘤、实体癌、产科灾难、严重肝功能衰竭、毒素和休克等疾病和状况发生。其中,对脓毒症和创伤病理生理的理解在DIC治疗中发挥了关键作用。在脓毒症相关的DIC中,及时诊断对于治疗的成功至关重要,包括早期抗生素治疗以治疗潜在原因,因为晚期DIC对治疗具有耐药性并且可能不可逆转。因此,脓毒症患者早期DIC的检测一直受到重视,并被归类为SIC(表1)。
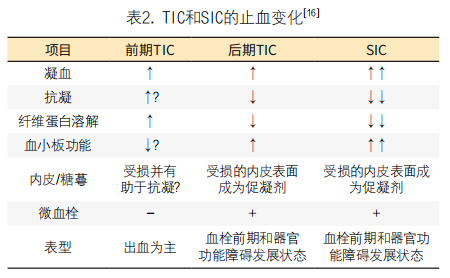
虽然TIC和SIC均符合显性DIC的诊断标准,但TIC的初始纤溶期(早期TIC)的表型与SIC不同。早期TIC存活的创伤患者可能在晚期(TIC晚期)出现血栓表型,表现为与SIC相似的多器官衰竭,也可能表现为大血管血栓性并发症,如深静脉血栓形成/肺栓塞。在这种情况下,凝血激活触发不平衡的纤溶,内皮功能受损和随后的微血栓形成在器官功能障碍的发展中起主要作用。然而,凝血激活、纤溶不平衡和内皮损伤在TIC和SIC中的致病意义往往不同。国际血栓与止血学会(ISTH)于2020年报道了SIC和TIC之间的异同,最近达成了共识[18]。
TIC和SIC在病理生理和表型上存在相当大的差异,但DIC的本质是全身激活和内皮损伤,这是两者的基础。TIC具有独特的特征,其特点是最初主要的止血功能障碍,并伴有高纤溶导致止血不充分。晚期TIC与SIC有器官功能障碍和血栓形成倾向的共同特征。现在有一个共识,TIC和SIC的通路至少部分重叠,但不完全相同。例如,严重出血常见于重大创伤,但在败血症中并不常见。另一方面,器官功能障碍是SIC的标志,这在TIC晚期也很常见。虽然抗凝血酶和重组凝血调节蛋白是有希望的SIC抗凝剂,但由于晚期TIC患病率较低,尚未对其效果进行研究。总之,虽然表型有很大的不同,但发病机制有重叠的特征。无菌和非无菌组织损伤通过多种机制系统性地激活凝血,凝血酶是两种情况下的关键因素。随后的内皮损伤、血小板功能受损和纤维蛋白溶解失调进一步加速组织缺血,最终导致危及生命的器官功能障碍和DIC。
健康的机体一定在各个方面处于精妙的平衡与调节控制过程中,出血与止血更是如此。检验专业要善于发现平衡、关键是要理解平衡。项目的开展与结果的解读更要贯彻与贯穿平衡!我们要有全局观和辩证观来解读相关检验结果与临床治疗和预后的关系,更好为临床服务,满足临床的要求,最终提升检验的学术地位。![]()
参考文献
K. M. and Ho, W. Pavey.Applying the cell-based coagulation model in the management of critical bleeding. Anaesth Intensive Care 2017 | 45: 2.
Smith SA. The cell-based model of coagulation. J Vet Emerg Crit Care (San Antonio) 2009; 19:3-10.
Cuker A, Siegal DM, Crowther MA, et al. Laboratory measurement of the anticoagulant activity of the non-vitamin K oral anticoagulants. J Am Coll Cardiol 2014; 64: 1128-1139.
Hoffman M, Monroe DM 3rd, Roberts HR. Activated factor VII activates factors IX and X on the surface of activated platelets: thoughts on the mechanism of action of high-dose activated factor VII. Blood Coagul Fibrinolysis 1998; 9 Suppl 1: S61-S65.
Maureane H, Dougald M. Monroe III. A Cell-based Model of Hemostasis. Thromb Haemost 2001; 85: 958–65.
Hoffman M, Monroe DM 3rd. A cell-based model of hemostasis. Thromb Haemost 2001; 85: 958-965.
Smith SA. The cell-based model of coagulation. J Vet Emerg Crit Care (San Antonio) 2009; 19: 3-10.
Perel P, Clayton T, Altman DG, Croft P, Douglas I, Hemingway H et al. Red blood cell transfusion and mortality in trauma patients: risk-stratified analysis of an observational study. PLoS Med 2014; 11: e1001664.
Murphy CH, Hess JR. Massive transfusion: red blood cell to plasma and platelet unit ratios for resuscitation of massive hemorrhage. Curr Opin Hematol 2015; 22: 533-539.
Volker Brinkmann and Arturo Zychlinsky.Beneficial suicide: why neutrophils die to make NETs. NATURE REVIEWS | MICROBIOLOGY VOLUME 5 | AUGUST 2007.
Vidal Delgado-Rizo, Marco A. Martínez-Guzmán, Liliana Iñiguez-Gutierrez, et,al.Neutrophil extracellular Traps and its implications in inflammation: An Overview.Frontiers in Immunology February 2017 | Volume 8 | Article 81.
Kimberly Martinod and Denisa D. Wagner. Thrombosis: tangled up in NETs.BLOOD, 1 MAY 2014 VOLUME 123, NUMBER 18.
Konstantin Stark and Steffen Massberg. Interplay between inflammation and thrombosis in cardiovascular pathology. Nature Reviews Cardiology, Vol. 18, Nr. 9: S. 666-682.
Anna S. ondracek , Irene M. Lang.Neutrophil Extracellular Traps as Prognostic Markers in COVID-19.Arterioscler Thromb Vasc Biol. 2021; 41: 995–998.
Bruna Gigante, Jerrold H. Levy, Eric van Gorp et, al. Management of patients on antithrombotic therapy with severe infections: a joint clinical consensus statement of the ESC Working Group on Thrombosis, the ESC Working Group on Atherosclerosis and Vascular Biology, and the International Society on Thrombosis and Haemostasis. European Heart Journal (2023) 00, 1–19.
Toshiaki Iba1, Julie Helms, Matthew D. Neal et, al. Mechanisms and management of the coagulopathy of trauma and sepsis: trauma-induced coagulopathy, sepsis-induced coagulopathy, and disseminated intravascular coagulation. J Thromb Haemost. 2023; 21: 3360–3370.
Iba T, Levy JH, Warkentin TE, Thachil J, van der Poll T, Levi M.Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J Thromb Haemost. 2019; 17: 1989–94.
Moore HB, Gando S, Iba T, et al. Defining trauma-induced coagulopathy with respect to future implications for patient management: communication from the SSC of the ISTH. J Thromb Haemost.2020; 18: 740-7.


